

Resumen
Las distrofias hereditarias de retina (DHR) son un grupo de enfermedades raras con una prevalencia de 1:3000-4000 personas. Afectan principalmente a los fotorreceptores de la retina y a las células epiteliales pigmentarias, y conducen a la neurodegeneración y finalmente a la apoptosis.
En 2021, publicamos los resultados del Hospital Universitario-Fundación Jiménez Díaz (Madrid, España), desde 1991 hasta agosto de 2019. Hemos actualizado estos resultados hasta agosto de 2022 realizando un estudio transversal retrospectivo de base hospitalaria sobre 4.794 familias no emparentadas afectadas por DHR, procedentes de todas las comunidades autónomas.
Las familias se clasificaron en: a) “NO-RP”: afectación predominante de conos, b) “RP” (retinosis pigmentaria): afectación primaria de bastones, y c) “DHR sindrómicas”: afectación visual junto con síntomas extraoculares. Los estudios moleculares incluyen: paneles dirigidos, exoma clínico y secuenciación exómica o genómica completa.
Se caracterizaron genéticamente el 62% (2962/4794) de las familias, y se identificaron 1.997 variantes (5.064 alelos) en 188 genes. El fenotipo más común fue RP (59%, 2465/4794). En cuanto al tipo de alelos, encontramos un 51% de missenses, 44% truncantes (nonsense, frameshift, indels y splicing) y 2% variaciones en el número de copias. Los genes mutados más frecuentemente fueron PRPH2, ABCA4 y RS1 en familias autosómicas dominantes (AD), autosómicas recesivas (AR) y ligadas al cromosoma X (XL) para las familias NO-RP, respectivamente; RHO, USH2A y RPGR en AD, AR y XL para RP; y MYO7A, USH2A y BBS1 en “DHR sindrómicas”. Las variantes patogénicas más frecuentes fueron ABCA4: p.Arg1129Leu y USH2A: p.Cys759Phe.
Nuestro estudio actualiza el sustrato genético y las características de las DHR en España, informando de la mayor cohorte presentada hasta la fecha y de un elevado número de genes causales implicados. Además, nuestros hallazgos tienen importantes implicaciones para el diagnóstico y el consejo genético, pero también para posibles opciones terapéuticas.Abstract
Inherited Retinal Dystrophies (IRDs) are a group of rare diseases with a prevalence of 1:3000-4000 people. They are genetic, primarily affecting retinal photoreceptors and epithelial pigmentary cells, and lead to neurodegeneration and finally apoptosis.
In 2021, we published our global results obtained in our registry at the Fundación Jiménez Díaz University Hospital (Madrid, Spain) from 1991 to August 2019. Now, we aimed to update these results until August 2022. Thus, we conducted a retrospective hospital-based cross-sectional study on 4.794 IRD-affected unrelated families from all the Spanish autonomous communities.
Families were classified into three phenotypic categories: a) “NON-RP” for cone-dominated phenotypes, b) “RP” (retinitis pigmentosa) for primary rod involvement, and c) “syndromic IRD” when visual plus extra-ocular symptoms are present. Molecular studies included: single-gene studies, clinical exome, whole exome or whole genome sequencing.
Overall, 62% (2962/4794) of the families were genetically characterized, in which 1.997 different likely causative variants (5.064 different alleles) were identified in 188 genes. The most common phenotype encountered was RP (59% of families, 2465/4794). Regarding the types of causative alleles, missenses were the most frequent (51%), followed by truncating (nonsense, frameshift, indels and splicing; 44%), while copy number variations were only 2%. The most recurrently mutated genes were PRPH2, ABCA4 and RS1 in autosomal dominant (AD), autosomal recessive (AR) and X-linked (XL) NON-RP families, respectively; RHO, USH2A and RPGR in AD, AR and XL for non-syndromic RP; and MYO7A, USH2A and BBS1 in syndromic IRD. Pathogenic variants ABCA4:p.Arg1129Leu and USH2A:p.Cys759Phe were the most frequent.
Our study provides the overall genetic landscape for IRD in Spain, reporting the largest cohort ever presented and a high number of causal genes involved in these diseases. Furthermore, our findings have important implications for genetic diagnosis and counseling, but also for possible therapeutic managementPalabras clave: Distrofias de Retina; Medicina Molecular; Genética Humana; Heterogeneidad Genética; Epidemiología Genética.
Keywords: Retinal Dystrophies; Molecular Medicine; Human Genetics; Genetic Heterogeneity; Genetic Epidemiology.
INTRODUCCIÓN
Las distrofias hereditarias de retina (DHR) son un conjunto de enfermedades raras (ER), producidas por la degeneración primaria de los fotorreceptores y/o el epitelio pigmentario de la retina (EPR), cursando con una gran heterogeneidad clínica y genética (1–3).
Cumplen los criterios de ER tanto por su prevalencia (1:3000-4000 personas) (4,5), como por sus características clínicas: crónicas, evolutivas y discapacitantes, pues conducen a baja visión y ceguera legal (2), con un gran impacto negativo en la calidad de vida y el ámbito emocional de personas afectadas y familiares (6).
Por sus manifestaciones oculares, las DHR pueden afectar principalmente a la retina periférica (p.ej.: retinosis pigmentaria (RP) o coroideremia), central o a ambas. En el caso de las retinopatías periférica, se afectan predominante los bastones, con ceguera nocturna, disminución del campo visual periférico y, finalmente, de la agudeza visual (7). Una forma periférica muy precoz es la Amaurosis Congénita de Leber (LCA) (8), caracterizada por nistagmo y una baja agudeza visual que es precoz y rápidamente progresiva. En las retinopatías centrales (p.ej.: distrofia de conos, acromatopsia o monocromatismo de conos azules) se afectan primariamente los conos, con fotofobia, alteración en la visión cromática y baja agudeza visual (9). De modo similar, en las distrofias maculares hereditarias (DM), se observa pérdida de visión central bilateral, acompañándose a menudo de atrofia macular y del EPR subyacente (p.ej.: enfermedad de Stargardt (STGD1, MIM#248200), distrofia macular viteliforme de Best (VMD2, MIM#153700). En casos de afectación mixta, como ocurre en la distrofia de conos y bastones (DCB) generalmente se produce en primer lugar la afectación central progresando a la periferia (9).
El 15%-20% de las DHR se asocian a patologías sistémicas (DHR sindrómicas) (2,7), con mayor discapacidad por la presencia de hipoacusia, discapacidad intelectual, malformación o neurodegeneración del sistema nervioso, alteraciones esqueléticas y/o trastornos endocrinológicos. La mortalidad de estas formas se incrementa como consecuencia de alteraciones metabólicas graves, malformaciones renales o cardiopatía (10,11).
El Síndrome de Usher es la forma sindrómica más frecuente con hipoacusia de moderada a profunda y, en algunos casos, alteración vestibular según los diferentes tipos clínicos tipo I (USH1, MIM#276900), tipo II (USH2, MIM#276901) y tipo III (USH3, MIM#276902). Otros síndromes frecuentes son el de Bardet-Biedl (BBS, MIM#209900) y Alström (ALMS, MIM#203800) (10,11).
Las DHR son muy heterogéneas genéticamente, con todos los patrones hereditarios mendelianos (herencia autosómica dominante, autosómica recesiva y ligada al X) y no mendelianos (mitocondrial, oligogénica, etc) descritos, así como más de 300 loci/genes asociados (https://web.sph.uth.edu/RetNet/sum-dis.htm, agosto 2022). Estos genes codifican proteínas implicadas en el desarrollo retiniano, mantenimiento de la estructura de los fotorreceptores, fototransducción y ciclo visual, procesos de fagocitosis, transporte nuclear, transcripción génica y splicing, entre otras funciones (2,3,12).
Gracias a las técnicas de secuenciación masiva (NGS, next-generation sequencing), en sus distintas aplicaciones de paneles dirigidos, exoma clínico (CES, clinical exome sequencing), exoma completo (WES, whole-exome sequencing) o genoma completo (WGS, whole-genome sequencing), la tasa de diagnóstico genético se ha incrementado significativamente, alcanzando valores del 50%-60% (12–17). El diagnóstico genético permite realizar diagnósticos diferenciales, así como un correcto asesoramiento familiar y, en algunos casos, establecer pronósticos. Además, ciertos casos ya pueden beneficiarse de tratamientos disponibles o futuros, o participar en ensayos clínicos.
El consorcio internacional para la investigación en ER (IRDiRC, https://irdirc.org/) tiene tres objetivos a alcanzar en 2027: i) acelerar un diagnóstico de calidad, ii) obtener tratamientos para un mayor número de ER y iii) evaluar el impacto que estas medidas tendrán en las vidas de pacientes/familias.
Hace tres décadas constituimos un grupo multidisciplinar orientado a la investigación traslacional de las DHR, denominado EsRetNet (Estudio Español de Retinosis Pigmentaria 1991-2006, https://www.esretnet.org/#). Este grupo ha trabajado en estrecha colaboración con investigadores españoles del CIBERER (Centro de investigación Biomédica en Red sobre ER, ISCIII https://www.ciberer.es/) y Raregenomics (Red de Investigación en Enfermedades Raras de la Comunidad de Madrid, https://www.rare-genomics.com/) e internacionales (ERDC, European Retinal Disease Consortium), adoptando similares objetivos a los de IRDiRC.
En concreto, nuestro objetivo fue la caracterización clínica y molecular de los pacientes con DHR en España, visibilizar sus necesidades y acercarles el diagnóstico y tratamiento.
En 2021, publicamos los resultados globales del estudio epidemiológico y genético en una gran cohorte de pacientes con DHR reunidos en un solo centro a lo largo del periodo 1991-2019 (18). En este artículo, realizamos una actualización de esos resultados en un corte transversal realizado en agosto de 2022.
MATERIAL Y MÉTODOS
Descripción de la cohorte
Se realizó un análisis retrospectivo de los pacientes con DHR de nuestro registro en el Hospital Universitario Fundación Jiménez Díaz (FJD, Madrid, España) desde 1991 hasta agosto de 2022, incluyendo todos los pacientes derivados al Servicio de Genética para diagnóstico genético por sospecha clínica de DHR.
Para el presente estudio, se han considerado los casos índices de cada familia incluida en el registro, excluyéndose aquellas sin análisis genético.
La cohorte completa contiene 4.794 casos índice de familias no relacionadas afectadas por DHR en nuestro registro, tras excluir aquellas sin análisis genético.
Este estudio fue realizado siguiendo todos los principios de la Declaración de Helsinki y posteriores revisiones, tras su aprobación por el Comité de Ética de la FJD (nº aprobación 134/2016_FJD) y la obtención de consentimiento informado escrito de todos los pacientes o de sus tutores legales.
Estudios clínicos
El examen clínico siguió criterios previamente descritos (18–20), e incluyó exámenes oftalmológicos, exploración física general y otras pruebas complementarias, así como datos de salud autoinformados.
Se revisaron los antecedentes clínicos y familiares a través de informes clínicos, un cuestionario específicamente diseñado para DHR y/o historia clínica electrónica de cada participante.
Clasificación clínica
La clasificación clínica inicial (diagnóstico “a priori”) o diagnóstico clínico-oftalmológico de presunción fue recogido en el momento de la solicitud del estudio genético, previo a los resultados. Está basado en los informes y datos clínicos remitidos en dicho momento.
De acuerdo con esos datos, y siguiendo criterios internacionales, las familias se clasificaron en 3 categorías de fenotipos, descritas previamente (7,18–22):
“NO-RP”: fenotipo con afectación predominante de conos: distrofia de conos, DCB, acromatopsia y monocromatismo de conos azules.
“RP”: fenotipo con afectación primaria de bastones: RP, LCA y coroideremia.
“DHR Sindrómicas”: cuando existen síntomas extraoculares.
Clasificación de fenotipos sindrómicos
Los fenotipos sindrómicos se clasificaron de acuerdo con los datos clínicos “a priori”, siguiendo criterios establecidos anteriormente por nuestro grupo (11,20). Para la categorización de los principales síntomas extraoculares se usaron términos de la ontología HPO (Human Phenotype Ontology) (23) y para las definiciones de los síndromes conocidos se utilizaron los criterios internacionales.
Clasificación del patrón hereditario
El patrón hereditario “a priori” se asignó en base al árbol genealógico, siguiendo criterios previamente publicados (24), dividiéndose en: autosómico dominante (AD), autosómico recesivo (AR), ligado al X (XL), esporádico (ES) y sin clasificar.
Estudio Molecular
A lo largo de los 32 años, se fueron aplicando diferentes abordajes de análisis moleculares, según la disponibilidad de la tecnología, como se ha publicado previamente (18).
A partir de 2013 (25) se incluyó la secuenciación masiva (NGS), inicialmente en el ámbito de la investigación, y en 2015 de modo rutinario el exoma clínico (CES) y, en casos seleccionados, la secuenciación exómica (WES) o genómica (WGS) completa.
Asimismo, se aplicaron distintos algoritmos bioinformáticos de análisis y reanálisis, para la anotación y priorización de las variantes genéticas (18,26,27).
Clasificación de las variantes y los genotipos
Las variantes genéticas se clasificaron según las guías internacionales del American College of Medical Genetics and Genomics (ACMG) (28) en 5 categorías: patogénicas, probablemente patogénicas, variantes de significado incierto (VUS), probablemente benignas y benignas.
A efectos de la caracterización genética de las familias, únicamente se consideraron las variantes patogénicas y probablemente patogénicas (clases 5 y 4).
Asimismo, en el presente trabajo únicamente se consideran como caracterizados aquellos casos con genotipos causales completamente identificados, no incluyéndose los casos con variantes monoalélicas para genes AR.
RESULTADOS
Clasificación clínica y de patrón hereditario
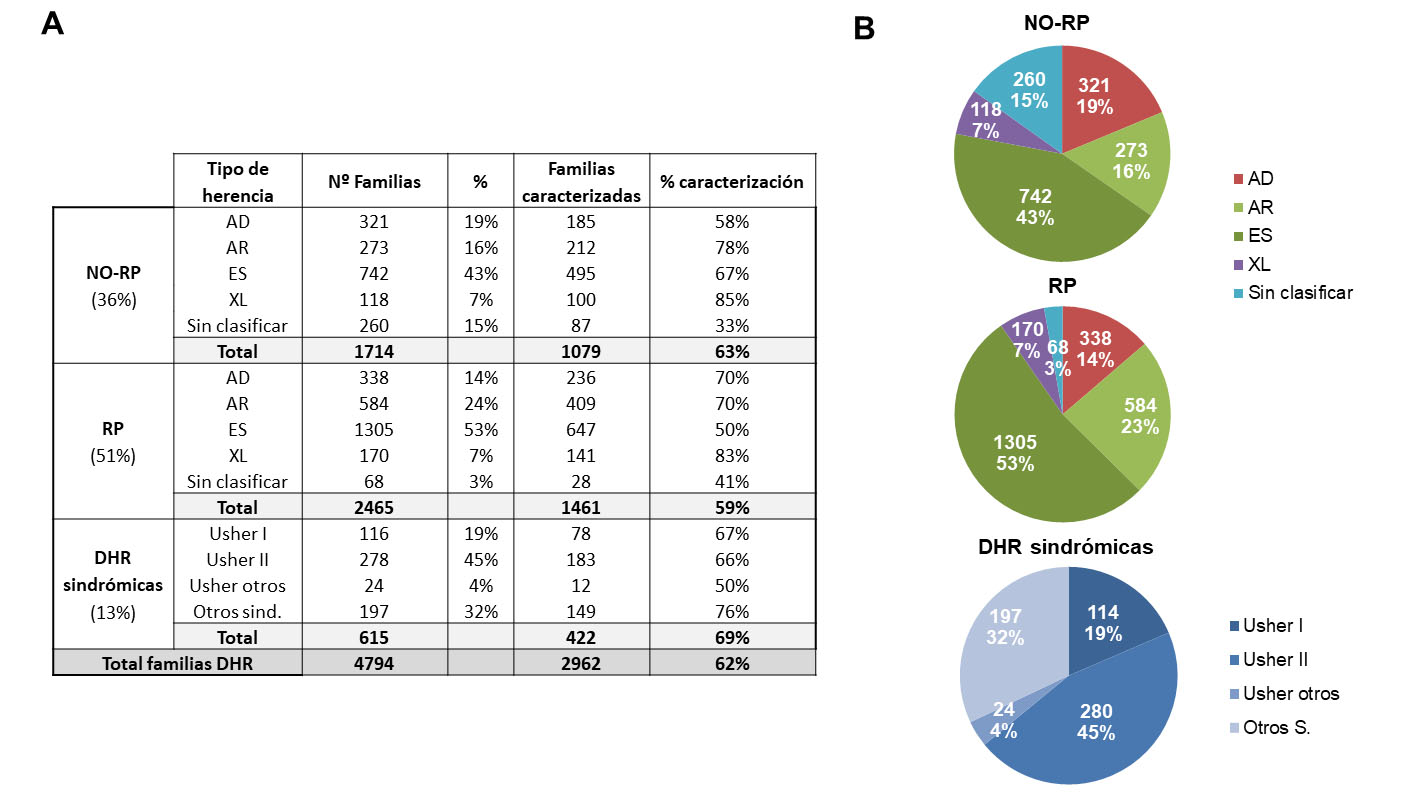
Las 4.794 familias DHR con estudios genéticos disponibles se clasificaron en tres categorías: “RP” (51% de las familias), “NO-RP” (36%) y formas sindrómicas (13%) (Figura 1A).
El 58% y 56% de las DHR no sindrómicas (“NO-RP” y “RP”) eran casos esporádicos (43% y 53%, respectivamente) o sin clasificar (15% y 3%, respectivamente). Entre los casos familiares, la herencia AR fue la más común, afectando al 16% (“NO-RP”) y 24% (“RP”) de las familias, seguida de las herencias AD y XL.
La herencia AR fue también la más común en las “DHR sindrómicas”, siendo el síndrome de Usher la forma sindrómica más prevalente (68%), representando el Usher tipo 2 el 45% del total de “DHR sindrómica” (Figura 1B).
Resultados de los estudios genéticos
Tasa diagnóstica y genes implicados
La tasa diagnóstica global fue del 62% (2962/4794 familias), y del 63%, 59% y 69% en los tipos “NO-RP”, “RP” y sindrómico, respectivamente (Figura 1).
Tras la caracterización molecular, se reclasificaron los 495 y 647 casos esporádicos, y 87 y 28 familias sin clasificar (“NO-RP” y “RP”, respectivamente).
Adicionalmente, se clasificaron correctamente el 4% (108/2540) de las familias “NO-RP” y “RP” (Figura 2A). Se comparó la herencia “a priori” basada en el árbol genealógico y la herencia final derivada del diagnóstico molecular (Figura 2B), obteniéndose que la mayoría de los casos esporádicos (495 “NO-RP” + 647 “RP”) tenían herencia AR después de las pruebas genéticas (“NO-RP”: 460/495, 93%; “RP”: 540/647, 84%).

Los casos esporádicos restantes se reclasificaron a AD (n=88) y XL (n=54), mientras 115 casos con modo de herencia desconocido inicialmente (87 “NO-RP” y 28 “RP”) se clasificaron como: AD (n=23), AR (n=81) y XL (n=11) tras los estudios.
En total se identificaron 188 genes y 1997 variantes distintas (5.064 alelos totales), causantes de DHR en las 2.962 familias diagnosticadas genéticamente (Figura 3).
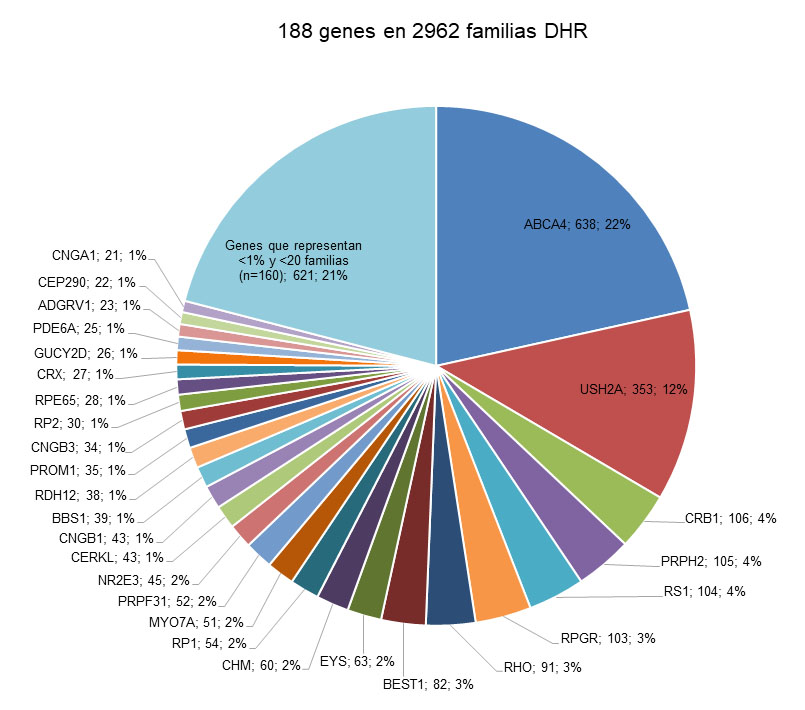
Aunque el número de genes implicados es elevado, solo 21 de ellos afectan a 30 o más familias (1%), mientras que los 167 restantes solo afectan a una (n=67) o a un número muy limitado de familias (Figura 3).
Los siete genes más frecuentemente implicados afectaron al 52% de las familias caracterizadas (ABCA4 (22%); USH2A (12%); CRB1 (4%); PRPH2 (4%); RS1 (4%); RPGR (3%) y RHO (3%)). Los tres primeros presentan mutaciones bialélicas y herencia AR; PRPH2 y RHO mutaciones en heterocigosis y una herencia AD; y RPGR una herencia XL.
En cuanto a la distribución de los genes causales por categorías clínicas (Figura 4A), en las formas “NO-RP” (63% de caracterización) se identificaron 73 genes. El gen ABCA4, responsable del 52% de las familias, junto con RS1, PRPH2 y BEST1, explican las tres cuartas partes de los casos índice “NO-RP” en España (Figura 4A-I). Por subtipos hereditarios, los genes más frecuentes fueron PRPH2, ABCA4 y RS1, para AD, AR y XL, respectivamente.

*Se indica la herencia y/o subtipo clínico finalmente considerado, tras los resultados genéticos.
Se indican subrayados aquellos genes que están presentes en más de un modelo hereditario y/o una categoría clínica. Entre paréntesis se indica el número de casos.
El grupo con la mayor heterogeneidad genética fue “RP”, con 119 genes causales, con 4 genes (USH2A, RPGR, RHO y ABCA4) afectando al 30% de las familias (Figura 4A-II). Por subtipos hereditarios, son RHO, USH2A y RPGR los más prevalentes en las familias AD, AR y XL, respectivamente.
En las formas sindrómicas, se identificaron 87 genes implicados, siendo los más frecuentes MYO7A, USH2A y BBS1, en el 56% de las familias “DHR sindrómicas” caracterizadas, los cuales son causantes de los síndromes de Usher de tipo 1, de tipo 2 y del síndrome de Bardet-Biedl, respectivamente (Figura 4A-III).
En cada subgrupo de la cohorte (pacientes “NO-RP” y “RP” divididos por modo de herencia) se observan algunos genes muy prevalentes (Figura 4B).
Algunos aparecen representados en más de una categoría, por transmitirse con dos posibles patrones de herencia (i.e.: PROM1, RP1: (AD y AR)), o por asociarse a distintos posibles fenotipos (USH2A: RP no sindrómica o síndrome de Usher tipo 2; PRPH2, RPGR: RP y NO-RP) (Figura 4B).
Mutaciones asociadas a DHR frecuentes en población española
Se ha observado una alta heterogeneidad genética en las DHR en nuestro país, con 5.064 variantes patogénicas causales observadas. Estas fueron mayoritariamente missense (51%), seguidas por las truncantes (nonsense, frameshift, indels y de splicing) (44%) y variaciones en el número de copias (CNV) (2%) (Figura 5A).
La Figura 5B muestra las 8 variantes más frecuentemente observadas en este estudio.
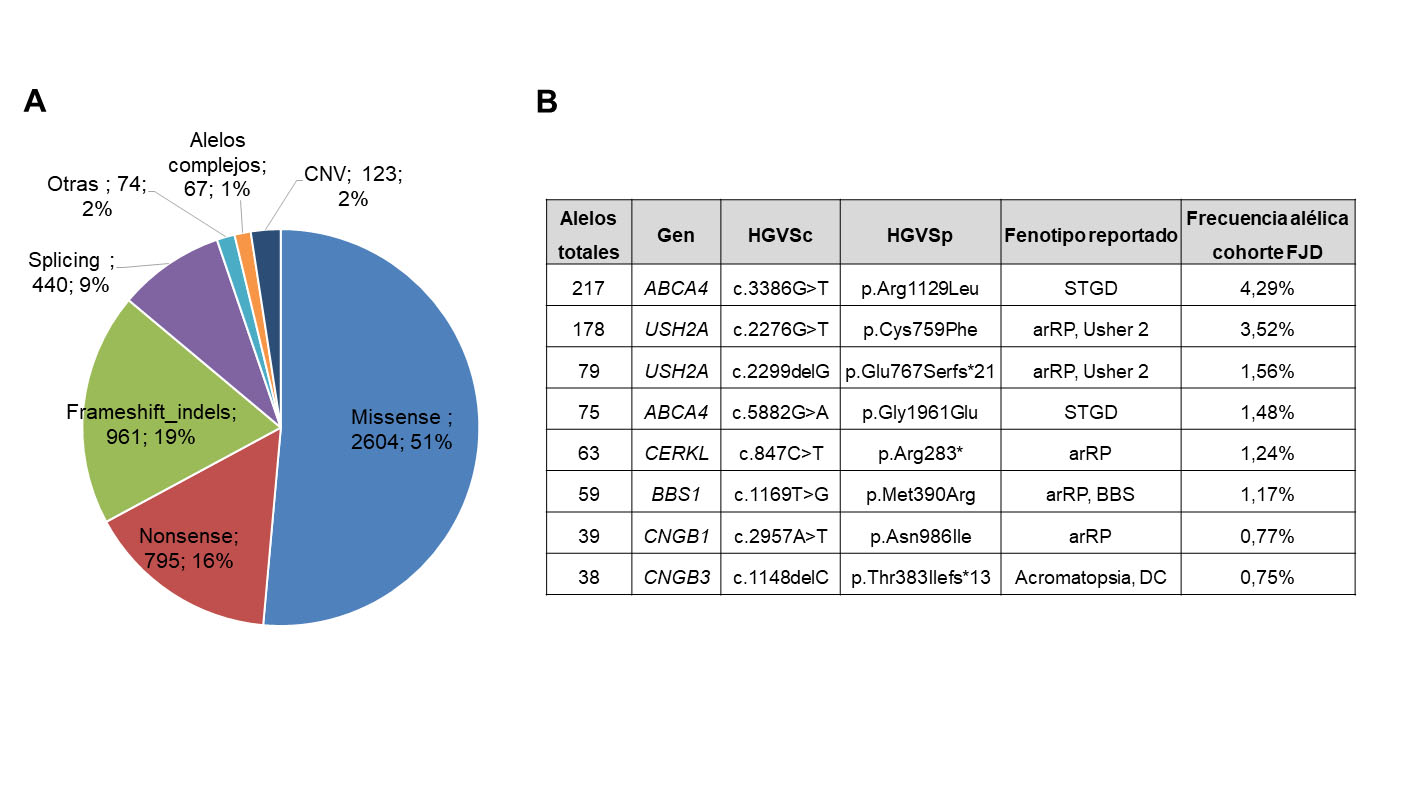
La más prevalente, ABCA4: p.Arg1129Leu (4,3% de alelos patogénicos identificados), afecta al 17,1% de las familias “NO-RP” caracterizadas. Se encuentra casi exclusivamente en pacientes españoles “NO-RP”, siendo con alta probabilidad una mutación fundadora española (29). La segunda variante más frecuente, USH2A: p.Cys759Phe (3,5% de los alelos) está presente en el 7,9% de las familias “RP”, además de en el 7,8% de las familias sindrómicas caracterizadas. Esta variante no es exclusiva de la población española y ha sido descrita también en otras poblaciones, siendo frecuente en Europa (30).
DISCUSIÓN
En los últimos años, se han publicado estudios de caracterización genética de DHR en distintas poblaciones (13–17,31–36), siendo el presente uno de los más grandes realizados (Tabla 1).

La eficacia diagnóstica en DHR varía en las distintas poblaciones (13–17,31–36), entre 37% (17) y 72% (32). Esta variabilidad es debida a distintos factores, como: las técnicas de estudio, obteniéndose mayor diagnóstico cuando se utilizan rutinariamente la NGS con análisis y reanálisis bioinformáticos, y con abordajes que incluyan un mayor número de genes o de regiones del genoma, como la WES, que incluye todas las regiones codificantes de todos los genes humanos, o la WGS, que incluye tanto regiones codificantes como no codificantes. Por otra parte, los métodos bioinformáticos que permiten analizar y anotar CNVs aportan una mayor tasa diagnóstica, que aquellos que solo anotan las variaciones de único nucleótido (SNV) y otras variantes como las pequeñas inserciones/deleciones.
En segundo lugar, dado que hay fenotipos clínicos con poca heterogeneidad genética (STGD, retinosquisis o coroideremia), la diferente composición de las cohortes en subtipos clínicos determina también la tasa diagnóstica (35).
Por otra parte, actualmente, se conocen unos 280 genes y 316 loci responsables de DHR (RetNet); lo que la convierte en una de las patologías mendelianas con mayor heterogeneidad descrita (5,36). Por ello, también el fondo genético puede contribuir a un mayor éxito en la tasa de genotipado en algunas poblaciones, bien por una baja variabilidad, por efecto cuello de botella (37), como ocurre en Reino Unido e Irlanda u otras regiones geográficamente aisladas, acumulándose ciertas mutaciones (16,36), o por tasas elevadas de consanguinidad (14).
Dada la elevada variabilidad genética, en España es necesario estudiar un alto número de genes para obtener tasas de diagnóstico comparables con las de otras poblaciones.
Así, en nuestro estudio se detectaron 188 genes, el número más elevado de las series consultadas (13–17,31–36) (Tabla 4), que reportan entre 18 (34) y 135 (16) genes que explican hasta el 74% de las familias estudiadas.
En un estudio reciente que describe el análisis de 185 genes causantes de DHR en 6 poblaciones distintas (5), se pone de manifiesto que hay tres genes (ABCA4, USH2A y EYS), cuyas mutaciones son muy frecuentes en al menos cinco de las seis subpoblaciones estudiadas. En nuestro estudio, los genes más mutados son también ABCA4 y USH2A, en el 22% y 12%, respectivamente de las familias afectadas (Figura 3), sin embargo, EYS solo explica el 2%.
Finalmente, la estructura genética de las distintas poblaciones se refleja en la existencia de mutaciones altamente prevalentes o exclusivas de población. Así Weisschuh et al. (35); reportan la variante p.Arg45Trp en RP1L1 como muy prevalente en Alemania, mientras que Whelan L. et al. (36) reportan como frecuente en Irlanda la variante c.4539+2028C>T en ABCA4. En nuestra cohorte, la variante p.Arg1129Leu en ABCA4 es la más prevalente (4,3% de los alelos encontrados) y parece ser exclusiva de población española, pues no aparece en otras series (38).
Un problema que urge abordar es identificar la causa genética en las familias sin caracterizar. Para ello es necesario aplicar herramientas recientemente desarrolladas como los nuevos algoritmos bioinformáticos junto con el abordaje mediante WGS de secuencias largas y cortas y otros estudios que permitan identificar reordenamientos genómicos y CNVs, así como acceder a regiones no bien caracterizadas: secuencias no codificantes (regiones reguladoras o intrónicas profundas), de secuencia repetida, o regiones intergénicas. Esta aproximación podría ayudar a revelar el componente genético oculto en los casos restantes. Por otra parte, estudios funcionales y análisis sobre las isoformas de los genes implicados en DHR que se expresan en la retina humana podrían arrojar luz sobre el papel de ciertas variantes genéticas en la etiopatogenia de la enfermedad.
Una consecuencia de la caracterización de los distintos subtipos de nuestra cohorte ha sido el genotipado en profundidad, facilitando estudios de correlación genotipo-fenotipo, y la eventual selección de subcohortes para posibles ensayos terapéuticos o tratamientos. Este abordaje se ha realizado exitosamente para la caracterización genética detallada de casos esporádicos (22), para la correlación clínica de DM y STGD (39), RPE65 (40), casos sindrómicos no Usher (11,20) y los tipos 1 y 2 del Síndrome de Usher (19), entre otros subgrupos.
En el caso particular de RPE65 estos estudios han permitido identificar los casos susceptibles de tratamiento rápidamente.
CONCLUSIONES
Nuestros resultados describen las características genéticas de los pacientes españoles con DHR, una de las series más grandes publicadas hasta ahora, indicando la gran heterogeneidad genética de esta enfermedad en una población, con elevado número de genes causantes implicados en la enfermedad.
La identificación de la causa genética en estos casos representó un paso clave para los pacientes. En primer lugar, permitió confirmar o reclasificar el subtipo clínico y hereditario, facilitando su asesoramiento genético, al determinar el riesgo en otros familiares y favorecer su prevención. En segundo lugar, es la condición necesaria para optar a terapias gen-dirigidas, bien ya autorizadas como la disponible para RPE65, o bien en el marco de ensayos clínicos.
En conclusión, este estudio actualiza el sustrato genético y las características de las DHR en España y ayudará a diseñar abordajes clínicos y asistenciales preventivos de este trastorno en nuestro país.
AGRADECIMIENTOS
Instituto de Salud Carlos III (PI16/00425, PI19/00321, PI19/00303, FI17/00192), CIBERER (06/07/0036), Biobanco IIS-FJD (PT13/0010/0012), Comunidad de Madrid (B2017/BMD-3721), FEDER, ONCE, Cátedra UAM-IIS-FJD de Medicina Genómica, Fundación Ramón Areces y Fundación Conchita Rábago, Proyecto UshTher (Programa Horizonte 2020 de la Unión Europea, Nº 754848).
Los autores agradecen a los restantes autores que aparecen mencionados en el manuscrito Perea-Romero et al., 2021 (PMID: 33972629) Rosario López‑Rodríguez, Lucía Pérez de Ayala, Elvira Rodríguez‑Pinilla, y a los pertenecientes a los grupos colaborativos “The ESRETNET Study Group”, “The ERDC Study Group”, “The Associated Clinical Study Group”, y a los pacientes que participaron en este estudio.
DECLARACIÓN DE TRANSPARENCIA
Los autores/as de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
BIBLIOGRAFÍA
- Wright AF, Chakarova CF, Abd El-Aziz MM, Bhattacharya SS. Photoreceptor degeneration: genetic and mechanistic dissection of a complex trait. Nat Rev Genet. 2010;11(4): 273-284.
- Ayuso C, Millán JM. Retinitis pigmentosa and allied conditions today: a paradigm of translational research. Genome Med. 2010; 2(5) :34.
- Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006; 368(9549): 1795-1809.
- Vaidya P. Retinitis pigmentosa: Disease encumbrance in the Eurozone. Int J Ophthalmol Clin Res [Internet]. 2015; 2(4). Disponible en: https://clinmedjournals.org/articles/ijocr/ijocr-2-030.php?jid=ijocr
- Hanany M, Rivolta C, Sharon D. Worldwide carrier frequency and genetic prevalence of autosomal recessive inherited retinal diseases. Proc Natl Acad Sci U S A. 2020; 117(5): 2710-2716.
- Cross N, van Steen C, Zegaoui Y, Satherley A, Angelillo L. Retinitis pigmentosa: Burden of disease and current unmet needs. Clin Ophthalmol. 2022; 16: 1993-2010.
- Verbakel SK, van Huet RAC, Boon CJF et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res. 2018; 66: 157-186.
- Kumaran N, Moore AT, Weleber RG, Michaelides M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Clinical features, molecular genetics and therapeutic interventions. Br J Ophthalmol. 2017; 101(9): 1147-1154.
- Gill JS, Georgiou M, Kalitzeos A, Moore AT, Michaelides M. Progressive cone and cone-rod dystrophies: Clinical features, molecular genetics and prospects for therapy. Br J Ophthalmol. 2019; 103(5): 711-720. doi: 10.1136/bjophthalmol-2018-313278.
- Millán JM, Aller E, Jaijo T, Blanco-Kelly F, Gimenez-Pardo A, Ayuso C. An update on the genetics of usher syndrome. J Ophthalmol. 2011; 2011: 417217.
- Perea-Romero I, Blanco-Kelly F, Sánchez-Navarro I et al. NGS and phenotypic ontology-based approaches increase the diagnostic yield in syndromic retinal diseases. Hum Genet. 2021; 140(12): 1665-1678.
- Farrar GJ, Carrigan M, Dockery A et al. Toward an elucidation of the molecular genetics of inherited retinal degenerations. Hum Mol Genet. 2017; 26(R1): R2-11.
- Huang XF, Huang F, Wu KC et al. Genotype–phenotype correlation and mutation spectrum in a large cohort of patients with inherited retinal dystrophy revealed by next-generation sequencing. Genet Med. 2015; 17(4): 271-278.
- Sharon D, Ben‐Yosef T, Goldenberg‐Cohen N et al. A nationwide genetic analysis of inherited retinal diseases in Israel as assessed by the Israeli inherited retinal disease consortium (IIRDC). Hum Mutat. 2020; 41(1): 140-149.
- Goetz KE, Reeves MJ, Gagadam S et al. Genetic testing for inherited eye conditions in over 6,000 individuals through the eyeGENE network. Am J Med Genet. 2020; 184(3): 828-837.
- Pontikos N, Arno G, Jurkute N et al. Genetic basis of inherited retinal disease in a molecularly characterized cohort of more than 3000 families from the United Kingdom. Ophthalmology. 2020; 127(10): 1384-1394.
- Colombo L, Maltese PE, Castori M et al. Molecular epidemiology in 591 Italian probands with nonsyndromic retinitis pigmentosa and usher syndrome. Invest Ophthalmol Vis Sci. 2021;62(2):13.
- Perea-Romero I, Gordo G, Iancu IF et al. Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications. Sci Rep. 2021; 11(1):1526.
- Blanco-Kelly F, Jaijo T, Aller E et al. Clinical aspects of Usher syndrome and the USH2A gene in a cohort of 433 patients. JAMA Ophthalmol. 2015; 133(2): 157-164.
- Sánchez-Navarro I, R J da Silva L, Blanco-Kelly F et al. Combining targeted panel-based resequencing and copy-number variation analysis for the diagnosis of inherited syndromic retinopathies and associated ciliopathies. Sci Rep. 2018; 8(1): 5285.
- Riveiro-Alvarez R, López-Martínez MA, Zernant J et al. Outcome of ABCA4 disease-associated alleles in autosomal recessive retinal dystrophies: Retrospective analysis in 420 Spanish families. Ophthalmology. 2013; 120(11): 2332-2337.
- Martín-Mérida I, Avila-Fernández A, Del Pozo-Valero M et al. Genomic landscape of sporadic retinitis pigmentosa: Findings from 877 Spanish cases. Ophthalmology. 2019; 126(8): 1181-1188.
- Köhler S, Schulz MH, Krawitz P et al. Clinical diagnostics in human genetics with semantic similarity searches in ontologies. Am J Hum Genet. 2009; 85(4): 457-464.
- Ayuso C, García-Sandoval B, Najera C, Valverde D, Carballo M, Antiñolo G. Retinitis pigmentosa in Spain. The Spanish Multicentric and Multidisciplinary Group for Research into Retinitis Pigmentosa. Clin Genet. 1995; 48(3): 120-122.
- Corton M, Nishiguchi KM, Avila-Fernández A et al. Exome sequencing of index patients with retinal dystrophies as a tool for molecular diagnosis. PLoS One. 2013; 8(6): e65574.
- Iancu IF, Perea-Romero I, Núñez-Moreno G et al. Aggregated genomic data as cohort-specific allelic frequencies can boost variants and genes prioritization in non-solved cases of inherited retinal dystrophies. IJMS. 2022; 23(15): 8431.
- Romero R, de la Fuente L, Del Pozo-Valero M et al. An evaluation of pipelines for DNA variant detection can guide a reanalysis protocol to increase the diagnostic ratio of genetic diseases. NPJ Genom Med. 2022; 7(1): 7.
- Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015; 17(5): 405-424.
- Riveiro-Alvarez R, Aguirre-Lamban J, López-Martínez MA et al. Frequency of ABCA4 mutations in 278 Spanish controls: an insight into the prevalence of autosomal recessive Stargardt disease. Brit J Ophthalmol. 2009; 93(10): 1359-1364.
- Reurink J, de Vrieze E, Li CHZ et al. Scrutinizing pathogenicity of the USH2A c.2276 G > T; p.(Cys759Phe) variant. NPJ Genom Med. 2022; 7(1): 37.
- Maeda A, Yoshida A, Kawai K et al. Development of a molecular diagnostic test for retinitis pigmentosa in the Japanese population. JPN J Ophthalmol. 2018; 62(4): 451-457.
- Motta FL, Martin RP, Filippelli-Silva R, Salles MV, Sallum JMF. Relative frequency of inherited retinal dystrophies in Brazil. Sci Rep. 2018; 8(1): 15939.
- Bravo-Gil N, González-del Pozo M, Martín-Sánchez M et al. Unravelling the genetic basis of simplex retinitis pigmentosa cases. Sci Rep. 2017; 7(1): 41937.
- Tiwari A, Bahr A, Bähr L et al. Next generation sequencing based identification of disease-associated mutations in Swiss patients with retinal dystrophies. Sci Rep. 2016; 6(1): 28755.
- Weisschuh N, Obermaier CD, Battke F et al. Genetic architecture of inherited retinal degeneration in Germany: a large cohort study from a single diagnostic center over a 9‐year period. Hum Mutat. 2020; 41(9): 1514-1527.
- Whelan L, Dockery A, Wynne N et al. Findings from a genotyping study of over 1000 people with inherited retinal disorders in Ireland. Genes. 2020; 11(1): 105.
- Chheda H, Palta P, Pirinen M et al. Whole-genome view of the consequences of a population bottleneck using 2926 genome sequences from Finland and United Kingdom. Eur J Hum Genet. 2017; 25(4): 477-484.
- Valverde D, Riveiro-Alvarez R, Bernal S et al. Microarray-based mutation analysis of the ABCA4 gene in Spanish patients with Stargardt disease: Evidence of a prevalent mutated allele. Mol Vis. 2006; 12: 902-908.
- Del Pozo-Valero M, Riveiro-Alvarez R, Blanco-Kelly F et al. Genotype–Phenotype correlations in a Spanish cohort of 506 families with biallelic abca4 pathogenic variants. Am J Ophthalmol. 2020; 219 :195-204.
- López-Rodríguez R, Lantero E, Blanco-Kelly F et al. RPE65-related retinal dystrophy: Mutational and phenotypic spectrum in 45 affected patients. Exp Eye Res. 2021; 212: 108761.
Carmen Ayuso
Departamento de Genética, Instituto de Investigación Sanitaria Hospital Universitario
Fundación Jiménez Díaz, Universidad Autónoma de Madrid (IIS-FJD, UAM), Madrid, España
Av. Reyes Católicos nº 2 · 28040 Madrid.
Tlf.: ++34 609 612 728 | E-Mail: cayuso@fjd.es
An RANM. 2022;139(03): 274-284
Enviado: 30.06.22
Revisado: 06.07.22
Aceptado: 12.07.22