

Resumen
Se ha observado la presencia de los ARN mensajeros del receptor de GLP-1 (receptor del péptido semejante al glucagón 1), GLUT-2 (isoforma 2 del transportador de glucosa) y GK (glucocinasa) en determinadas áreas cerebrales, lo cual podría relacionarse con el proceso sensor de glucosa recientemente descrito en las células β pancreáticas. Tanto GLUT-2, GK, como la proteína reguladora de la glucocinasa (GKRP) han sido identificadas en cerebros de humanos y rata. La expresión del gen de la GK da lugar a una proteína de 52 kDa, caracterizada por métodos bioquímicos e inmunocitoquímicos, que tiene una alta afinidad y una alta capacidad fosforilante de la glucosa en hipotálamo y corteza cerebral. Su importancia fisiológica viene dada porque se activa cuando se eleva la glucemia por la ingesta de alimentos. Además la localización subcelular de GK y GKRP facilita el proceso sensor de glucosa controlado por GK y GKRP, las cuales podrían contribuir a la génesis de una sensación de saciedad. Esta observación junto con la actividad anorexígenica del GLP-1 sugieren además un papel conjunto de este péptido sobre el control de la ingesta de nutrientes. Sería interesante conocer si las mutaciones de los genes de las proteínas antes citadas producen alteraciones patológicas, así como si las modificaciones de la conducta alimentaria pueden estar relacionadas con la capacidad sensora de glucosa. Hasta ahora nos hemos centrado en el papel de la GK como sensor de glucosa en el hipotálamo y su relación con la conducta alimentaria, pero no debemos olvidar que esta enzima se encuentra ampliamente expresada en corteza cerebral, donde podría desempeñar importantes funciones.
Abstract
The presence of the messenger RNA for GLP-1 receptor (GLP-1R), GLUT-2 (isoform 2 of glucose transporter ) and GK (glucokinase) in brain areas might be related to the glucose sensing process that was recently described in pancreatic β cells. GLUT-2, GK and the regulatory protein of GK (GKRP) were identified in both human and rat brains. The GK gene expression generates a 52 kDa protein, identified by biochemical and immunochemical methods; it shows a high Km for glucose and a high capacity to phosphorylate glucose in the hypothalamus and in the brain cortex. Its physiological importance is related to its activation secondary to an increase in glycaemia related to food ingestion. GK and GKRP modulate the enzymatic activity according to the metabolic needs of the cells. This is necessary for the sensing process that is controlled by GK and GKRP, and could also contribute to a sensation of satiety. It would be interesting to know if mutations in the genes codifying the mentioned proteins can produce pathological alterations, or whether changes in the feeding behavior are related to the brain glucose sensing process. Until now, our work has focused on the role of hypothalamic glucokinase as a glucose sensor related to food behavior, but we cannot forget that this enzyme is widely expressed in the brain cortex, where it could also carry out other important functions. These observations, together with the anorexigenic activity of GLP-1 (glucagon-like peptide-1), suggest a combined role of these peptides on food intake control.
Palabras clave: Cerebro; Glucocinasa; Sensor glucosa; Control; Conducta alimentaria.
Keywords: Brain; Glucokinase; Glucose sensing; Control; Food behaviour.
INTRODUCCIÓN
Como es sabido el cerebro es el órgano central que controla las actividades de otras estructuras, así como la génesis del pensamiento, inteligencia, aprendizaje, sensaciones y metabolismo, junto con la coordinación de la conducta y movimientos entre otras funciones. Para llevar cabo estas actividades el cerebro, que posee el 2% del peso corporal, utiliza el 20% de la glucosa corporal y consume alrededor del 25% del oxígeno total. También tiene una escasa capacidad para almacenar glucosa y otros nutrientes por lo que necesita recibirlos de forma continuada para la realización de sus funciones y para su propia supervivencia; la interrupción del flujo sanguíneo produce daños tisulares irreparables y la muerte (Tabla 1). A pesar de ser el órgano más importante su conocimiento ha permanecido estancado hasta las últimas décadas.

Por otra parte el cerebro necesita gran cantidad de energía para realizar sus diversas funciones, tales como el mantenimiento de las concentraciones iónicas a través de las membranas de las neuronas, la generación de los potenciales de acción, y el funcionamiento de los canales iónicos necesarios para la transmisión de los impulsos nerviosos (1,2,3,4). La glucosa es la principal fuente de energía del cerebro, en el que penetra a través de la barrera hematoencefálica (BBB), vía difusión facilitada mediante transportadores de glucosa, proporcionando la energía en forma de ATP necesaria para las funciones de las neuronas y células de glia.
También otras hormonas producidas en el intestino como el GLP-1 (péptido semejante al glucagón 1) y el GIP (péptido inhibidor gástrico) con acción incretina, así como la hormona pancreática amilina están asociadas con el metabolismo de la glucosa (5,6). Otras biomoléculas como la leptina, resistina y adiponectina están relacionadas con la homeostasis de la glucosa. Alteraciones de los valores normoglucémicos tienen efectos deletéreos que aumentan la morbilidad y mortalidad de la población.
Con objeto de evitar marcadas oscilaciones de la glucosa circulante, sensores de ella están localizados en intestino, células endocrinas pancreáticas, vena porta, sistema nervioso central y células neuroendocrinas (3,4,5,6), lo que permite generar los mecanismos necesarios para mantener sus concentraciones fisiológicas.
CAPACIDAD SENSORA DE GLUCOSA EN CEREBRO
En el estudio que realizamos para la identificación y caracterización de la GK como sensor cerebral de glucosa (1), tuvimos unos antecedentes previos con el análisis del papel del receptor de GLP-1 sobre la conducta alimentaria, que nos orientaron hacia las conclusiones finales. En efecto mediante la técnica de la hibridación in situ encontramos la colocalización de los ARN mensajeros de los receptores de GLP-1, GK y GLUT-2 en la pared del tercer ventrículo, núcleo arqueado, eminencia media y núcleo supraóptico (7,8). Estas áreas cerebrales contienen neuronas sensoras de glucosa responsables de la conducta alimentaria. Además estas proteínas han sido relacionadas con el proceso sensor de glucosa en las células β pancreáticas, lo que sugería por una parte un papel del GLP-1 en la regulación hipotalámica de la ingestión de macronutrientes y por otra el papel de GK y GLUT-2 como moléculas sensoras de glucosa. En efecto encontramos que el GLP-1 se comportaba como un péptido anorexígeno (8) mientras que con la GK y el GLUT-2 iniciamos las investigaciones encaminadas a identificar y caracterizar la enzima de origen cerebral respecto a las previamente descritas en hígado e islotes pancreáticos.
La expresión génica de la GK cerebral de rata da lugar a una proteína de 52 kDa con, una alta Km para la glucosa, sin inhibición por la glucosa-6-fosfato y una contribución a la actividad fosforilante de la glucosa total entre el 40 y el 14%, siendo el hipotálamo y la corteza cerebral las regiones de máxima actividad (9). Estos hallazgos sugerían que la GK presente en el cerebro de rata podría facilitar la adaptación de este órgano a las fluctuaciones de las concentraciones de la glucosa sanguínea, a la vez que la expresión de GK y GLUT-2 en las mismas células hipotalámicas podrían indicar un papel sensor de glucosa.
Demostrada la existencia de GK en cerebro de humanos y ratas (2, 9), fundamentalmente con características similares a las presentes en hígado y células pancreáticas β, pudimos incluirlas a todas como isoenzimas pertenecientes a la familia de las hexocinasas que, catalizan la fosforilación de la glucosa a glucosa-6-fosfato. Las hexocinasas I,II y III tienen una alta afinidad por la glucosa, con baja Km, que son inhibidas por glucosa-6-fosfato. La GK o hexocinasa IV tiene una baja afinidad por la glucosa y no es inhibida por las concentraciones fisiológicas de la glucosa-6-fosfato (8), lo cual explica su importancia fisiológica ya que se activa cuando los niveles circulantes de glucosa aumentan tras la ingesta de alimentos mientras que la falta de inhibición por glucosa-6-fosfato hace posible que la hexosa metabolizada sea proporcional a la presente en el espacio extracelular.
La existencia de promotores tejido específicos de la enzima permiten una regulación diferencial por un tipo de promotor en hígado y otro diferente en cerebro y las células β pancreáticas (9,10). Asimismo las concentraciones de GK en cerebro y células β pancreáticas son controladas por la glucosa mientras que la insulina es el efector de la GK en hígado.
Pero la capacidad de la GK como molécula sensora de glucosa no sólo reside en la posesión de una escasa afinidad por la glucosa y por no ser inhibida por la glucosa-6-fosfato, sino también por la existencia de una proteína reguladora de la GK (GKRP) presente en hígado, células β pancreáticas y ahora descrita por nosotros en cerebro (11), como lo prueban las bandas obtenidas por inmunotransferencia de GK y GKRP presentes en las fracciones subcelulares de hígado e hipotálamo, así como las imágenes obtenidas por técnicas inmunocitoquímicas en islotes pancreáticos, hipotálamo y corteza cerebral nuclear que une y transporta la GK al núcleo (11).
Hallazgos similares fueron encontrados por nosotros en cerebros humanos gracias a la excelente colaboración del Dr. Alberto Rábano (2). En efecto los ARN mensajeros y las proteínas GKRP, GK y GLUT-2 fueron identificados en varias regiones cerebrales.Todo ello hace posible que el aumento de la glucemia después de las comidas pueda ser detectado por neuronas específicas en el hipotálamo debido a las altas Km de GLUT-2 y GK (12,13,14).
En resumen nuestros resultados indican que la GK junto la GKRP presentes en el cerebro de humanos y rata realizan un papel sensor de glucosa (15,16), que podrían jugar un importante papel en la conducta alimentaria, contribuyendo a la génesis de una sensación de saciedad (17).
Cuando las neuronas son expuestas a grandes concentraciones de glucosa, la activación de la GK produce un incremento del cociente ATP/AMP, inactivándose el canal K ATP. Con ello se produce una despolarización de la membrana (15), que inducela entrada de calcio dependiente del voltaje.
OTROS SENSORES METABÓLICOS
Además de las propiedades sensoras de la GK, GKRP y GLUT-2 citadas con anterioridad también son conocidas las importantes funciones que realizan la proteína cinasa activada por ATP (AMPK), la diana en mamíferos de la rapamicina (mTOR) y la cinasa PAS (PASK).
La AMPK es un complejo heterotrimérico que tiene un dominio serina/treonina que reconoce la energía celular mediante la detección del cociente AMP/ATP (18), que es activada en estados con escasa carga energética o estrés metabólico que disminuyen la producción de ATP. Cuando ella se activa estimula las rutas catabólicas como la glucolísis y la β-oxidación de los ácidos grasos e inhibe las vías anabólicas responsables de la síntesis de macromoléculas recuperando de esta forma el balance energético a nivel central y periférico (19). También la AMPK hipotalámica está implicada en la conducta alimentaria y en la termogénesis del tejido adiposo marrón (20,21).
El complejo mTORC1 es una serina/treonina cinasa que forma parte de la ruta mTOR/S6K, que es activada por aminoácidos, mitógenos, factores de crecimiento y estados energéticos favorables que promocionan los estados anabólicos, mientras que los estados de estrés celular y depleción energética suprimen dicha ruta (22). También este complejo es un sensor energético relacionado con la ingesta de alimentos y el control del peso corporal (23,24). Tanto el mTORC1 y el AMPK participan juntos en la regulación de la ingesta de alimentos, aunque una cantidad reducida de nutrientes estimula AMPK pero sin cambios en el complejo mTORC1. Estos hallazgos muestran que estos sensores pueden participar de diferente manera en el control de la conducta alimentaria (25).
PAS CINASA (PASK)
Se trata de la proteína que posee un dominio N terminal Per-Arnt-Sim (PAS) y un dominio catalítico C terminal serina/treonina cinasa (26). Como AMPK y mTORC1 es una proteína que responde a nutrientes y regula el metabolismo de la glucosa y la energía celular, que también responde a una variedad de señales intracelulares, entre las que se encuentran el oxígeno, el estado redox y la luz entre otras muchas (27). En mamíferos PASK puede regular la síntesis de glucógeno, metabolismo energético y la traducción de proteínas (28,29,30) así como está relacionada con la epigenética y la diferenciación(31). También PASK es importante para mantener los efectos sobre los nutrientes de AMPK y mTORC1/S6K1 hipotalámicos (32,33,34) y para la regulación sobre el sensor de la glucosa GK (35) y el transductor insulínico Akt. Por otra parte la deficiencia PASK evita muchos de los efectos perjudiciales de las dietas ricas en grasas sobre el hígado (36) y, por ello disminuyen los depósitos grasos. Asimismo la deficiencia de PASK disminuye la expresión de varios factores de transcripción que estimulan la producción de enzimas gluconeogénicas (35). También la deficiencia de PASK evita la resistencia a la acción de la insulina (37,38) y la intolerancia a la glucosa que se manifiesta durante el envejecimiento (39) lo que previene la disminución de varias enzimas antioxidantes relacionadas con la edad (40).
CONDUCTA ALIMENTARIA Y CAPACIDAD SENSORA DE GLUCOSA EN CEREBRO
Sin duda el desarrollo de los proyectos de investigación hacen posible el avance de nuestro conocimiento sobre un determinado tema pero a su vez plantean nuevas interrogantes, que posiblemente están relacionados con la limitada información inicial de los tópicos que tratamos y con la aparición de nuevos hallazgos. En este orden de ideas la existencia de la GK y la GKRP descritas por nosotros en cerebro junto con las proteínas presentes en los canales K+ATP de las neuronas GR y GS plantean la posibilidad de mutaciones que puedan expresar alteraciones patológicas. Recordemos que esas modificaciones en las células β pancreáticas pueden producir diabetes o en otros casos generan situaciones con incrementos en la secreción de insulina.
Dado que propusimos en su día que la GK cerebral podría estar implicada en el control de la ingesta de los alimentos y recientemente se ha demostrado su papel en el control de la saciedad (17), sería de interés conocer si las mutaciones de los genes de las proteínas antes citadas podrían ser responsables de algunas patologías aún no definidas completamente.
En la misma dirección podría ser de interés conocer si entidades nosológicas implicadas con alteraciones de la conducta alimentaria tales como la anorexia nerviosa, bulimia, obesidad o diabetes podrían estar relacionadas con alteraciones de la capacidad sensora de glucosa en cerebro.
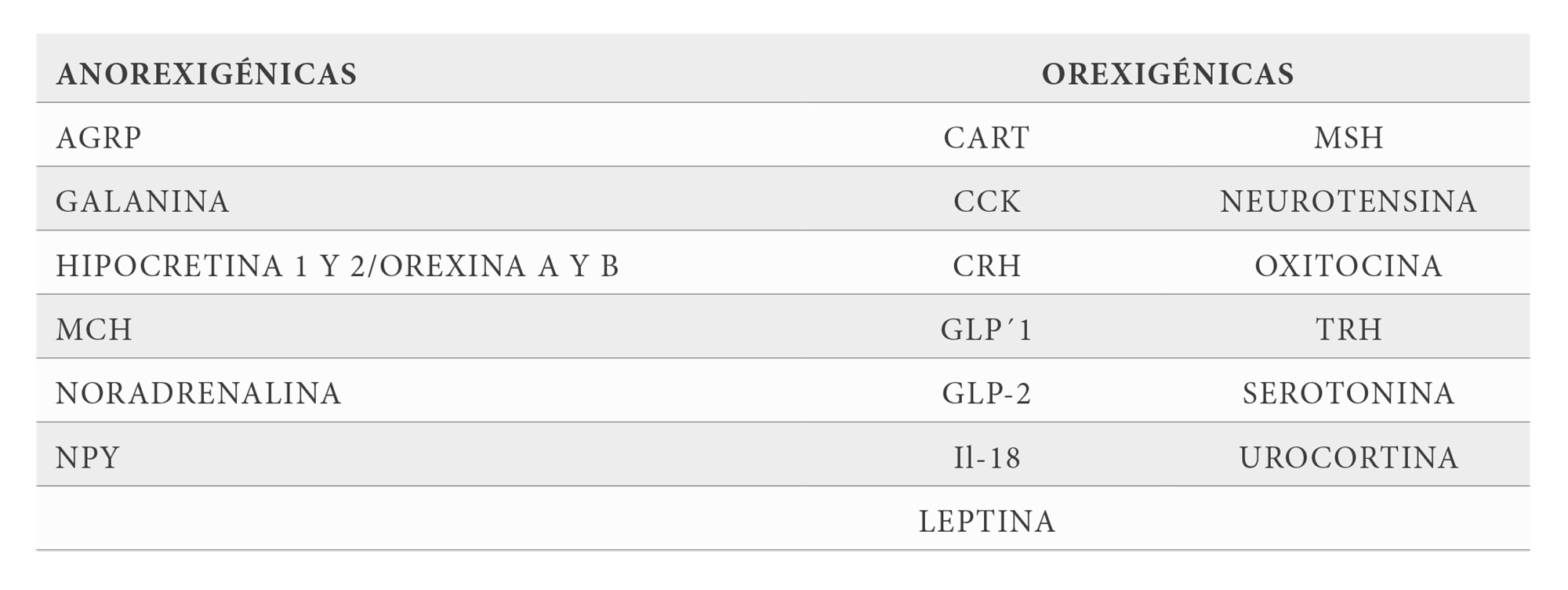
Desde antiguo se ha propuesto que las concentraciones circulantes de glucosa pueden ser necesarias para controlar la ingesta de los alimentos. A principios del siglo XX A. Carlson (41) sugirió que las bajas concentraciones de glucosa estimulan la toma de alimentos mientras que las elevadas concentraciones de la hexosa podrían facilitar la terminación de la ingesta. Más tarde en 1953 Mayer propuso la hipótesis glucostatica (42) en la que postuló que el aumento de la glucemia por la ingesta de alimentos activaría las neuronas hipotalámicas responsables del proceso de terminación. Desde entonces hasta ahora se ha producido un avance excepcional de los aspectos moleculares, celulares y fisiológicos de la ingesta de los alimentos y sus implicaciones fisiopatológicas. Dentro de este relato conviene destacar el papel del hipotálamo en este proceso, especialmente mediante sus núcleos ventromedial (VMN), arqueado (AN) y área lateral hipotalámica (LHA) entre otros, así como los tanicitos (43) que revisten el tercer ventrículo y son capaces de recoger la información que llega desde el exterior por esta vía y los agentes orexígenicos y anorexígenicos (Tabla 2) y sus receptores, que permiten a GK, GKRP y transportadores de glucosa contribuir al control de la conducta alimentaria.
Además de los componentes tisulares citados existen otros muchos que participan en la génesis de la conducta alimentaria y su relación con la capacidad sensora de la glucosa en cerebro, entre los cuales podemos citar en primer lugar a los tanicitos, células de glia situadas en la pared del tercer ventrículo, bañadas por el CSF que las relaciona con otras áreas cerebrales y que mediante sus prolongaciones interaccionan con neuronas localizadas en los núcleos hipotalámicos (43). Ambos tipos celulares participan en el metabolismo de la glucosa, de forma que los tanicitos a través de la vía glucolítica transforman la glucosa en lactato y este es transferido por los transportadores monocarboxilados (MCT) hasta las neuronas donde completan el proceso oxidativo de la glucosa. Se han descrito hasta 14 isoformas de MCT, de los cuales las isoformas MCT -1,-2 y -4 están relacionados con el tema que nos ocupa (44,45). También en los tanicitos hay receptores para los agentes orexígenicos y anorexígenicos, así como GK,GKRP y GLUT-2, lo que sugiere que pueden intervenir en el proceso sensor de glucosa y de activación de los mecanismos estimulantes o inhibidores de la ingesta de alimentos. En efecto en las paredes del tercer ventrículo cerebral se encuentran los receptores de GLP-1, GK y GKRP, sugiriendo que ellas pueden intervenir en las manifestaciones anorexígenicas del péptido, de la misma forma que ocurre para otras moléculas.
Aparte de la importancia que tienen GLUT-2, GK y GKRP como moléculas sensoras de glucosa existen otros transportadores de glucosa y hexocinasas de baja Km y alta afinidad por la glucosa. Nos referimos a los otros GLUTs y a los cotransportadores (46,47) de sodio y glucosa (SGLTs). En cuanto a estos últimos se ha descrito un posible mecanismo sensor de glucosa no metabólico en el hipotálamo, en el que las neuronas GR son activadas por la elevación de las concentraciones de glucosa mediante la corriente generada por el cotransporte de dos sodio y una glucosa por SGLTs.
En cuanto las formas predominantes en cerebro de las isoformas GLUTs, el GLUT-1 está localizado en las células de glia, en las células endoteliales de la barrera hematoencefálica (BBB) y en los α y β1tanicitos, mientras que el GLUT-3 está situado en las neuronas (48). Además la alta afinidad por la glucosa de estos dos transportadores de glucosa y las hexocinasas de baja Km por la glucosa están en consonancia con la menor concentración de la hexosa en cerebro que en otros tejidos, lo que permite un apropiado uso de nutrientes. También la elevada constante catalítica (Kcat)) de GLUT-3 respecto a GLUT-1 hace posible que GLT-3 pueda responder asimismo a altas concentraciones de glucosa (49).
El elevado número de moléculas que participan de forma directa o relacionada con el proceso de metabolización de glucosa en cerebro bajo condiciones normales, abre la puerta a sugerencias sobre las posibles alteraciones de ellas en situaciones patológicas. Hasta ahora nos hemos centrado en el papel de la GK como sensor cerebral de glucosa en el hipotálamo y la conducta alimentaria pero, no olvidamos que esta enzima se encuentra también ampliamente expresada en otras áreas cerebrales, especialmente en la corteza, donde podría desempeñar importantes funciones.
AGRADECIMIENTOS
Actualmente la actividad científica se desarrolla en equipo o incluso con la colaboración entre grupos de trabajo con objeto de obtener un mayor avance de los temas tratados. Por ello quiero expresar mi más profundo agradecimiento a todos los miembros de nuestro grupo de investigación y a los de otros grupos con los que colaboramos, por su activa participación a lo largo de años y cuya participación ha sido ampliamente reconocida en la Bibliografía de esta publicación. También este proyecto de investigación fue posible gracias a la generosa contribución del Ministerio de Investigación e Innovación, Fondo de Investigación Sanitaria, Instituto de Salud Carlos III, Madrid y las Fundaciones Ramón Areces, Mutua Madrileña y Eugenio Rodríguez Pascual, Madrid.
DECLARACIÓN DE TRANSPARENCIA
El autor/a de este artículo declara no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
BIBLIOGRAFÍA
- ↑Matschinsky FM. Perspectives in diabetes: Glucokinase as a glucose sensor and metabolic signal generator in pancreatic β-cells and hepatocytes. Diabetes. 1990; 39(6): 647-652.
- ↑Roncero I, Álvarez E, Chowen JA et al. Expression of glucose transporter isoform GLUT-2 and glucokinase genes in human brain. J Neurochem. 2004; 88: 123-1210.
- ↑Burcelin R, Da Costa A, Drucker D, Thorens B. Glucose competence of the hepatoportal vein sensor requires the presence of an activated glucagón-like peptide-1 receptor. Diabetes. 2001; 50: 1720-1728.
- ↑Jetton TL, Liang Y, Petetepher CC et al. Analysis of upstream glucokinase promoter activity in transgenic mice and identification of glucokinase in rare neuroendocrine cells in the brain and gut. J Biol Chem. 1994; 269: 3641-3654.
- ↑Barneo J, Pardal R, López-Barneo J. Low glucose-sensing cells in the carotid body. Nat Neurosci. 2002; 5: 197-198.
- ↑Liu M. Semo S, Kirchgessner AL. Identification and characterization of glucoresponsive neurons in the enteric nervous system. J Neurosci. 1999; 19: 10305-10317.
- ↑Álvarez E, Roncero I, Chowen JA, Thorens B, Blázquez E. Expression of the glucagon-like peptide-1 receptor gene in rat brain. J Neurochem. 1996; 66: 920-927.
- ↑Navarro M, Rodríguez de Fonseca F, Álvarez E et al. Colocalization of glucagon-like peptide-1 (GLP-1) receptors, glucose transporter GLUT-2, and glucokinase mRNAs in rat hypothalamic cells: Evidence for a role of GLP-1 receptor agonists as an inhibitory signal for food and water intake. J Neurochem. 1996; 67: 1982-1991.
- ↑Roncero I, Álvarez E, Vázquez P, Blázquez E. Functional glucokinase isoforms are expressed in rat brain. J Neurochem. 2000; 74: 1848-1857.
- ↑Magnuson MA, Shelton KD. An alternative promoter in the glucokinase gene is active in the pancreatic β cell. J Biol Chem. 1989; 264: 15936-15942.
- ↑Álvarez E, Roncero I, Chowen JA, Vázquez P, Blázquez E. Evidence that glucokinase regulatory protein is expressed and intreacts with glucokinase in rat brain. J Neurochem. 2002; 80: 45-53.
- ↑Dean PM, Mathews EK, Sakamoto Y. Pancretic islet cells: Effects of monosaccharides, glycolytic intermediates and metabolic inhibitors on membrane potential and electric activity. J Physiol. 1975; 246: 456-478.
- ↑Yang XJ, Kow LM, Funabashi T, Mobbs CV. Hypothalamic glucose sensor: Similarities to and differences from pancreatic beta-cell mechanisms. Diabetes. 1999; 18: 1763-1772.
- ↑Yang XJ, Kow LM, Pfaff DW, Mobbs C V. Metabolic pathways that mediate inhibition of hypothalamic neurons by glucose. Diabetes. 2004; 53: 67-73.
- ↑Karschin C, Aschcroft FM, Karschin A. Overlapping distibution of KATP channel-forming unit Kir6.2 subunit and the sulfonylurea receptor SUR 1 in rodent brain. FEBS Lett. 1997; 401: 9-64.
- ↑Dunn-Meynell AA, Routh VH, Kang I, Gaspess L, Levin BF. Glucokinase is the likely mediator of glucose sensing in both glucose-excited and glucose-inhibited central neurons. Diabetes. 2002; 5: 2056-2065.
- ↑Uranga RM, Millán C, Barahona MJ et al. Adenovirus-mediated supression of hypothalamic glucokinase affects feeding behavior. Sci Rep. 2017; 7: 3697. https://doi.org/10.1038/s41598-017-03928-x
- ↑García D, Shaw RJ. AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell. 2017; 66: 789-800.
- ↑Hardie DG. AMP-activated protein kinase: Maintaining energy homeostasis at the cellular and whole-body levels. Annu Rev Nutr. 2014; 14: 31-55.
- ↑López M. Hypothalamic AMPK and energy balance. Eur J Clin Invest. 2018; 48(9): e12996.
- ↑Andersson U, Filipsson K, Abbott CR et al. AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem. 2004; 279: 12005-12008.
- ↑Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017; 169(2): 361-371.
- ↑Cota D, Proulx K, Smith KA et al. Hypothalamic mTOR signaling regulates food intake. Science. 2006; 312( 5775): 927-930.
- ↑Hu F, Xu Y, Liu F. Hypothalamic roles of mTOR complex 1: Integration of nutrient and hormone signals to regulate energic homeostasis. Am J Physiol Endocrinol Metab. 2016; 310(11): E994-E1002.
- ↑Dagon Y, Hur E, Zhen B, Wellenstein K, Cantley LC, Kahn BB. P70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin´s. Effect on food intake. Cell Metab. 2012; 16: 104-112.
- ↑Kikani CK, Antoyisamy SA, Bonanno JB et al. Structural bases of PAS domain regulated kinase (PASK) activation in the absence of activation loop phosphorylation. J Biol Chem. 2010; 285(52): 41034-41043.
- ↑Moglich A, Ayers RA, Moffat K. Structure and signaling mechanism of Per-ARNT-Sim domains. Structure. 2009; 17: 1282-1294.
- ↑DeMille D, Grose JH. PAS kinase: a nutrient sensing regulator of glucose homeostasis. IUBMB Life. 2013; 65(11): 921-929.
- ↑Zhang DD, Zhang JG, Wang YZ, Liu GL, Liu XY. Per-Arnt-Sim kinase (PASK): an emerging regulator of mammalian glucose and lipid metabolism. Nutrients. 2015; 7: 7437-7450.
- ↑Grose JH, Rutter J. The role of PAS kinase in PASsing the glucose signal. Sensors. 2010; 10: 5668-5682.
- ↑Hurtado-Carneiro V, Pérez García A, Álvarez E, Sanz C. PAS Kinase: a nutrient and energy sensor “master key” in the response to fasting/feeding conditions. Front Endocrinol. 2020; 11: 594053.
- ↑Hurtado-Carneiro V, Roncero I, Blázquez E, Álvarez E, Sanz C. PAS kinase as a nutrient sensor in neuroblastoma and hypothalamic cells required for the normal expression and activity of other cellular nutrients and energy sensors. Mol Neurobiol. 2013; 48(3): 904-920.
- ↑Hurtado-Carneiro V, Roncero I, Egger SS et al. PAS kinase is a nutrient and energy sensor in hypothalamic areas required for the normal function of AMPK and mTOR/S6K1. Mol Neurobiol. 2014; 50 (2): 314-326.
- ↑Hurtado-Carneiro V, Sanz C, Roncero I, Vázquez P, Blázquez E, Álvarez E. Glucagon-like peptide 1 (GLP-1) can reverse AMP- Activated Protein Kinase (AMPK) and S6 Kinase (P70S6K) Induced by fluctuations in glucose levels in hypothalamic areas involved in feeding behaviour. Mol Neurobiol. 2012; 45: 348-361.
- ↑Pérez-García A, Dongil P, Hurtado-Carneiro V, Blázquez E, Sanz C, Álvarez E. PAS kinase deficiency alters the glucokinase function and hepatic metabolism. Sci Rep. 2018; 8(1): 1091.
- ↑Pérez-García A, Dongil P, Hurtado-Carneiro V, Blázquez E, Sanz C, Álvarez E. High-fat diet alters PAS kinase regulation by fasting and feeding in liver. J Nutr Biochem. 2018; 57: 14-25.
- ↑Blázquez E, Velázquez E, Hurtado-Carneiro V, Blázquez E, Ruiz-Albusac JM. Insulin in the brain: Its pathophysiological implications for states related with central insulin resistance, type 2 diabetes and alzheimer´s disease. Front Endocrinol. Neuroendocrine Sci. 2014; 5(161): 2-22
- ↑Blázquez E, Hurtado-Carneiro V, LeBaut-Ayuso Y et al. Significance of brain glucose hypometabolism, altered insulin signal transduction, and insulin resistance in several neurological diseases. Front Endocrinol. Neuroendocrine Sci. 2022; 13. https://doi.org/10.3389/fendo.2022.873301
- ↑Dongil P, Pérez-García A, Hurtado-Carneiro V, Herrero-de-Dios C, Álvarez E, Sanz C. PAS kinase deficiency reduces aging effects in mice. Aging (Albany NY). 2020; 12(3): 2275-2301.
- ↑Dongil P, Pérez-García A, Hurtado-Carneiro V et al. PAS kinase deficiency triggers antioxidant mechanisms in the liver. Sci Rep. 2018; 8(1): 13810.
- ↑Carlson AJ. The control of hunger in health and disease. Chicago IL:The University of Chicago Press; 1919.
- ↑Mayer J. Glucostatic mechanism of regulation of food intake. N Engl J Med. 1953; 249(1): 13-16.
- ↑Elizando-Vega R, Cortes-Campos Ch, Barahona MJ, Oyarce KA, Carril CA, García.Robles MA. The role of tanycytes in glucosensing. J Cell Mol. 2015; 19(7): 1471-14821.
- ↑Carneiro L, Pellerín L. Monocarboxylate transporters: new players in body weight regulation. Obes Rev. 2015; 16: 55-66.
- ↑Halestrap AP, Price WT. The protein-linked monocarboxylated transporter (MCT) family: Structure, function and regulation. Biochem J. 1999; 343: 281-299.
- ↑Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev. 2011; 91: 733- 794.
- ↑O´Malley B, Reimann F, Simpson AK et al. Sodium-coupled glucose cotransporters contribute to hypothalamic glucose sensing. Diabetes. 2006; 55: 3381-3386.
- ↑McEwen BS, Reagan LP. Glucose transporter expression in the central nervous system: Relationship to synaptic function. Eur J Pharmacol. 2004; 450: 13-24.
- ↑Simpson IA, Carruthers A, Vannucci SJ. Supply and demand in cerebral energy metabolism: The role of nutrient transporters. J Cereb Blood Flow Metab. 2007; 27: 1766-1791.
ranm tv
Enrique Blázquez Fernández
Real Academia Nacional de Medicina de España
C/ Arrieta, 12 · 28013 Madrid
Tlf.: +34 91 547 03 18 | Email de correspondencia
Enviado*: 31.05.22
Revisado: 12.06.22
Aceptado: 20.07.22
* Fecha de lectura en la RANM