

Resumen
Desde el feto hasta la adolescencia, el crecimiento físico es un fenómeno biológico complejo que se encuentra determinado por múltiples factores genéticos, epigenéticos, genómicos, ambientales y nutricionales. El patrón de crecimiento prenatal es claramente diferente al postnatal. Mientras que el primero no depende de la acción de hormona de crecimiento, el segundo, depende de la acción de esta hormona, conjuntamente con la tiroxina, los esteroides sexuales y la nutrición. Aún en la actualidad, la definición del hipocrecimiento es compleja, debiendo establecerse la normalidad de una talla dada para la edad, sexo y etnia del individuo. Según se mantengan o no las proporciones normales entre los distintos segmentos corporales el hipocrecimiento se considera armónico o disarmónico. De enorme interés es la aproximación al diagnóstico molecular de las alteraciones del crecimiento, habiendo aprendido mucho durante los últimos veinte años de las bases moleculares de la “deficiencia aislada de hormona de crecimiento”, deficiencia combinada de hormonas hipofisarias”, “resistencia a la acción de la hormona de crecimiento”, “deficiencia de IGF-I”, resistencia a IGF-I, deficiencia de IGF-II, anomalías en el gen de la proteína transportadora ácido lábil y, más recientemente de la deficiencia de la proteasa PAPP-A2.
Estos elementos se analizarán en detalle en este manuscrito.
Abstract
From fetal life to adolescence, human growth is a dynamic biological phenomenon and a good indicator of health. This complex phenomenon is influenced by genetic, hormonal, nutritional and environmental factors, both pre – and postnatally. While prenatal growth is growth hormone independent, postnatally it depends on the action of this hormone, as well as thyroxine, sex steroids and nutrition. The concept of short stature remains complex even today and its normality must be established according to height for age, sex and ethnia of the subject. Short stature can be proportionate or not. Of great interest is the molecular approach for the diagnosis of growth abnormalities. In fact, we have learned much during the last twenty years on the molecular basis of growth hormone deficiency, combined deficiency of pituitary hormones, resistance to growth hormone, IGF-I deficiency, resistance to IGF-I, IGF-II deficiency, primary IGFALS deficiency and, more recently, deficiency of the protease PAPP-A2.
These elements will be analyzed in this manuscript.
Palabras clave: Crecimiento; Prenatal; Postnatal; Hormona de crecimiento; Factores de crecimiento semejantes a la insulina; Proteína ácido lábil; Proteasas.
Keywords: Growth; Prenatal Postnatal; Growth hormone; Insulin-like growth factors; Acid labile protein; Proteases.
INTRODUCCIÓN
El crecimiento físico y la maduración de un niño se encuentran determinados por múltiples mecanismos genéticos, epigenéticos y genómicos, conjuntamente con factores ambientales, ya intrínsecos, ya extrínsecos (1,2).
Aún en la actualidad, la definición del hipocrecimiento es compleja, debiendo establecerse la normalidad de una talla dada para la edad, sexo y etnia del individuo. No obstante, como quiera que presenta un componente genético, también debe estudiarse en su propio contexto familiar. Grosso modo, se compara la talla adulta potencial de un sujeto con su “talla genética”. Junto a ello, es menester valorar de forma sensible la velocidad de crecimiento (ganancia de talla en cm/año cronológico). Todas estas dificultades metodológicas son las responsables de que no sea posible establecer un punto de corte que discrimine de forma nítida entre tallas “normales” y “patológicas” y de que no exista un consenso internacional que establezca con claridad el concepto de talla baja/hipocrecimiento.
En la práctica clínica, se suelen utilizar como criterios de hipocrecimiento los que siguen:
- Las tallas que se sitúan por debajo de – 2 SDS (percentil 2,3) para la edad, sexo y etnia del sujeto (≈ percentil 3/ – 1,88 SDS).
- Las tallas que aun estando entre ± 2 SDS para la población general, se sitúan más de 2 SDS por debajo del carril de crecimiento correspondiente a su talla genética
- Predicción de talla adulta: más de 2 SDS por debajo de la talla genética.
- Una velocidad de crecimiento que se mantiene persistentemente disminuida.
Al igual que ocurre con la talla, no existe un consenso que establezca con claridad qué es una velocidad de crecimiento disminuida, aunque suele considerarse como “potencialmente” patológica, cuando es inferior a – 1 SDS (≈ percentil 25) para la edad y sexo, pero mantenida durante más de 2-3 años.
En la práctica clínica, el hipocrecimiento patológico es la manifestación de muchas patologías, constituyendo un verdadero Tratado de Pediatría el establecimiento de un diagnóstico preciso.
Según se mantengan o no las proporciones normales entre los distintos segmentos corporales el hipocrecimiento se considera armónico o disarmónico. Los hipocrecimientos disarmónicos representan un mínimo porcentaje de los hipocrecimientos y en su mayoría son de base genética (osteocondrodisplasias).
Mayor complejidad supone el enfrentamiento al diagnóstico molecular de las diferentes enfermedades pediátricas y endocrinológicas que se han ido descubriendo en los últimos veinte años: deficiencia aislada de hormona de crecimiento, deficiencia combinada de hormonas hipofisarias, resistencia a la acción de la hormona de crecimiento, alteraciones en el gen IGF-I y su receptor, alteraciones en el gen de IGF-II y anomalías en el gen de la proteína transportadora ácido lábil del complejo ternario (IGF-I, IGFBP-3 y ALS) circulante, cuya excisión por proteasas permitirá a los factores de crecimiento ejercer o no su acción sobre el cartílago epifisario generando el crecimiento físico de forma adecuada o anómala (2).
INCIDENCIA
El establecimiento de la incidencia o prevalencia de los hipocrecimiento en un momento dado es ciertamente complejo (3). Si se considera aisladamente el concepto estadístico de talla baja (< – 2 SDS), el 2,3 % de la población entraría en el concepto hipocrecimiento. No obstante, el número de niños que se miden en los estudios poblacionales es insuficiente para determinar con fidelidad los extremos de la curva de Gauss que representa la distribución de la talla y, dado que es más factible que el crecimiento se vea severamente frenado que fuertemente acelerado, es probable que el porcentaje de tallas bajas sea mayor del 2,3 %. Por último, en el concepto de hipocrecimiento se incluyen otros criterios, por lo que, puede presumirse que, el número de niños con hipocrecimiento en un momento dado sería superior al 3-5 % de la población infantil.
ETIOPATOGENIA Y CLASIFICACIÓN
Dentro de los hipocrecimientos, clásicamente, se han diferenciado dos grandes grupos etiopatogénicos: los hipocrecimientos considerados como normales o “variantes normales de talla baja” (VNTB) y los hipocrecimientos patológicos. En la actualidad (Tabla 1), tienden más a clasificarse como hipocrecimientos de causa conocida o desconocida (talla baja idiopática – TBI-) (4,5)
3.1. Hipocrecimientos de causa conocida
Representan el 20 % del total, siendo la consecuencia de trastornos patológicos que alteran la capacidad de crecimiento intrínseca de los tejidos (osteocondrodisplasias, alteraciones del metabolismo óseo, retrasos de crecimiento intrauterino, cromosomopatías y síndromes dismórficos), sus mecanismos reguladores (hipocrecimientos de causa endocrinológica) o el ambiente interno (malnutrición y patología crónica en diferentes órganos y sistemas) y emocional del niño (hipocrecimiento psicosocial).
Desde un punto de vista etiopatogénico y diagnóstico (Tabla 1), es útil tener en consideración el momento de su inicio (pre o postnatal) y si se conservan o no las proporciones corporales normales (armónicos o disarmónicos). Los hipocrecimientos de inicio prenatal (6) suelen ser el resultado de una agresión al ambiente fetal o de un defecto genético; por el contrario, los de inicio postnatal suelen responder a una agresión iniciada fuera del período de vida intrauterino. En lo que se refiere al mantenimiento o no de la armonía corporal, los hipocrecimientos disarmónicos son siempre patológicos; mientras que, los armónicos pueden ser normales o patológicos.

3.1.1. Hipocrecimientos de inicio prenatal
Se considera a un recién nacido (RN) como pequeño para su edad gestacional (RNPEG), cuando su peso (PRN) y/o longitud (LRN) al nacimiento se encuentran, al menos 2 SDS por debajo de la media para su edad gestacional (≤ – 2 SDS) (7). En los países desarrollados, sólo el 4-7 % de los RN son RNPEG; sin embargo, es una de las causas más importantes de talla baja, ya que este antecedente se encuentra en el 20% de los adultos con talla baja.
El patrón de crecimiento de los RNPEG suele ser característico. En efecto, alrededor del 80-90 % experimentan un crecimiento de recuperación, parcial o total, durante el primer o segundo año de vida y alcanzan una talla dentro de la normalidad (± 2 SDS). Por el contrario, el 10-20 % restante mantiene la talla baja después de los 2 años y en el 50 % de éstos la talla final será baja. La edad ósea (EO) suele estar retrasada, pese a lo cual la pubertad se inicia habitualmente a una edad normal o incluso ligeramente adelantada, alcanzando una talla final baja, que en la mayoría de los casos es similar en SDS a la talla prepuberal.
Una tercera parte de los RNPEG se deben a factores fetales (cromosomopatías, anomalías congénitas y síndromes dismórficos) y las otras dos terceras partes a factores maternos (malnutrición, infecciones y tóxicos, entre otros) y uterino-placentarios (malformaciones uterinas o arteria umbilical única, entre otras). Hasta en un 40 % de los casos no se identifica ninguna causa patológica. En general y en comparación con los RN de peso adecuado a su edad gestacional, los RNPEG presentan una elevada morbimortalidad perinatal, así como una mayor frecuencia de secuelas a largo plazo, como son, entre otras: disminución del rendimiento intelectual y psicológico, hipocrecimiento postnatal y desarrollo de anomalías hormonales (pubarquia prematura e hiperandrogenismo ovárico) y metabólicas en la edad adulta (resistencia a la insulina, obesidad, diabetes mellitus tipo 2, dislipemia, hipertensión arterial y enfermedad cardiovascular) (7).
3.1.1.1. RNPEG por factores fetales
Multitud de cuadros sindrómicos (Russell-Silver, Seckel, Cornelia de Lange, Noonan o Prader-Willi, entre otros) (8-10), al igual que la mayoría de las cromosomopatías, presentan talla baja, con frecuencia de inicio prenatal. Son frecuentes también, los rasgos sindrómicos, las malformaciones en diferentes órganos y un grado variable de retraso mental.
Entre las cromosomopatías, merecen especial mención por su frecuencia: el síndrome de Down (trisomía 21; 1:600 RN vivos) y el síndrome de Turner (45,X0 y sus variantes; 1:2.500-3.000 RN vivas). Las características fenotípicas del síndrome de Down facilitan su diagnóstico neonatal; por el contrario, el diagnóstico del síndrome de Turner suele hacerse más tardíamente, debido a que sus rasgos síndrómicos característicos pueden ser poco manifiestos y pasar desapercibidos. En el síndrome de Turner, el fracaso de crecimiento, sobre todo a partir de los 2-3 años (sólo un 16 % son RNPEG) es prácticamente constante (11).
3.1.1.2. RNPEG por factores uterinos y placentarios
Existe una clara asociación entre el retraso del crecimiento fetal y la disfunción placentaria o las anomalías uterinas y del cordón; ya que, la disminución del tamaño de la placenta, la reducción del área de intercambio útil (placenta baja, desprendimiento placentario) o su perfusión insuficiente (arteria umbilical única, hemangioma placentario) pueden determinar una disminución del aporte de nutrientes y de oxígeno para el feto, condicionando así sus posibilidades de crecimiento.
3.1.1.3. RNPEG por factores maternos
Dentro de los factores maternos, la desnutrición materna crónica es la causa más frecuente de RNPEG en los países subdesarrollados. En los países del llamado primer mundo, su contribución al nacimiento de RNPEG es difícil de establecer, ya que suele ir asociada a otros factores también vinculados con el nacimiento de RNPEG.
3.1.2. Osteocondrodisplasias
Las osteocondrodisplasias representan anomalías primarias del hueso y del cartílago de base genética y herencia variable. Individualmente son entidades raras, pero colectivamente integran un grupo numeroso (2-5:10.000 RN) de 436 entidades nosológicas diferentes, distribuidas en 42 grupos, con un número de genes de 364 (12) (Tabla 2).
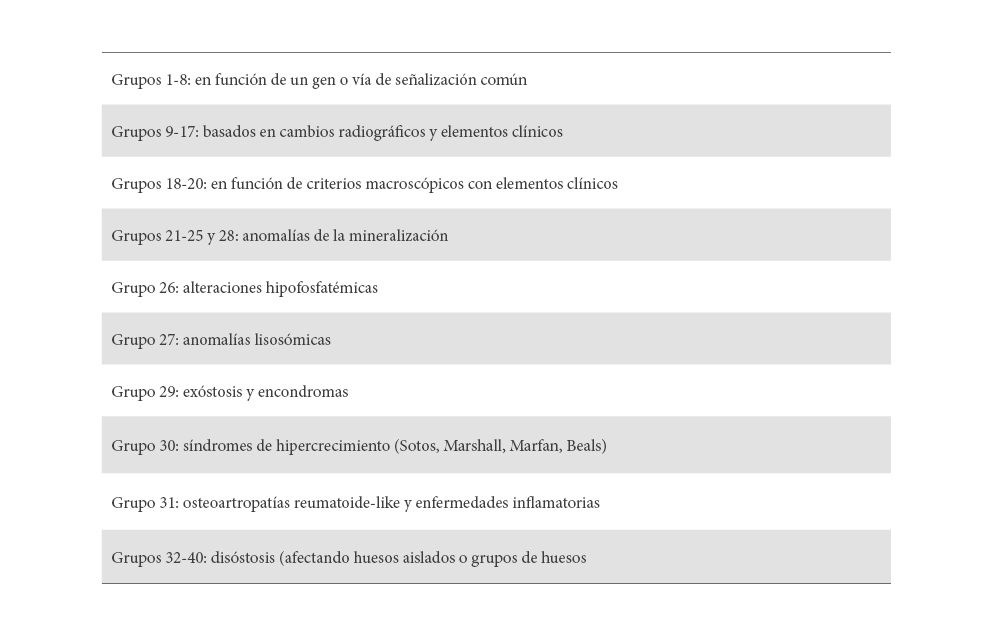
El hipocrecimiento es habitualmente disarmónico; sin embargo, en algunos casos la disarmonía corporal o no existe o es tan poco manifiesta que pasan con facilidad a formar parte del diagnostico diferencial de la talla baja idiopática. Además, debe tenerse en cuenta que en la clasificación más reciente, algunos cuadros clínicos de hipercrecimiento se incluyen dentro de las displasias óseas.
Entre las osteocondrodisplasias que por su frecuencia, expresividad clínica, herencia dominante y patrón de crecimiento pueden confundirse con un hipocrecimiento armónico, especialmente con una variante normal de talla baja, se encontrarían la hipocondroplasia, la pseudoacondroplasia, las displasias epifisarias múltiples (autosómicas dominantes y autosómicas recesivas) y la haploinsuficiencia o insuficiencia del gen SHOX (short stature homeobox-containing gene), que incluye el síndrome de Turner, la discondrosteosis de Léri-Weill y la displasia mesomélica de Langer (12).
3.1.2.1. Hipocondroplasia (MIM 146000)
Se ha descrito como una “forma leve” de acondroplasia, cursando con cambios esqueléticos cualitativamente semejantes a ésta última, pero cuantitativamente más modestos. Ambas enfermedades se heredan de forma autosómica dominante y se deben a mutaciones en el mismo gen (FGFR3; 4p16.3). La mutación responsable más frecuente es Asn540Lys, pero sus bases moleculares son más heterogéneas que en el caso de la acondroplasia y otros genes podrían estar implicados (12).
Los signos clínicos más relevantes, incluyen:
- Talla baja disarmónica, con tronco y extremidades relativamente cortos.
- Cráneo normal o ligeramente aumentado.
- Ligera lordosis lumbar.
Los signos radiológicos más comunes son los que siguen:
- Acortamiento de huesos largos.
- Acortamiento y ensanchamiento del cuello femoral.
- Alargamiento de la parte distal del peroné con respecto a la tibia.
- Ausencia de mano en tridente tan evidente como en la acondroplasia.
⇒ Con estos signos clínicos y radiológicos hay que proceder al estudio molecular del gen FGFR3. Recientemente, se han mostrado mutaciones en el gen SHOX en algún paciente con hipocondroplasia. Se estima que el 60% de los pacientes con hipocondroplasia tienen una transversión del nucleótido 1620 C1620A en el gen FGFR3, dando lugar a una sustitución ASN540Lys (N540K).
3.1.2.2. Pseudoacondroplasia (MIM 177170)
Los signos clínicos más relevantes, incluyen (12):
- Talla baja disarmónica, miembros cortos, con proporciones corporales similares a la acondroplasia, pero con cráneo y facies normales.
- Tronco desproporcionadamente alargado con lordosis lumbar marcada y escoliosis moderada en algunos pacientes.
- Genu valgum, Genu varum o deformidades irregulares.
- Hipermotilidad de todas las articulaciones, excepto los codos.
Los signos radiológicos más relevantes son los que siguen:
- Aplanamiento moderado de los cuerpos vertebrales con deformidad biconvexa.
- Irregularidades de las epífisis femorales.
- Acortamiento de huesos largos de las manos, con metáfisis ensanchadas.
- Acortamiento de huesos largos, con metáfisis marcadamente irregulares y epífisis deformadas.
⇒ Con estos signos clínicos y radiológicos hay que proceder al estudio molecular del gen COMP (19p13.1-p12), que codifica la proteína de la matriz oligomérica del cartílago.
3.1.2.3. Displasias epifisarias múltiples –autosómicas dominantes-(MIM 132400, 600204, 600969, 607078)
Se han descrito varios tipos de displasia epifisaria múltiple con un patrón de herencia autosómico dominante y con mutaciones en diferentes genes (COMP –tipo 1, COL9A1 –tipo 6-, COL9A2 –tipo 2-, COL9A3 –tipo 4 – y MATN3 –tipo 5-). Junto a ello, se ha descrito una forma con patrón de herencia autosómico recesivo debida a mutaciones en el gen DTDST (MIM 226900) (12).
Los signos clínicos más relevantes, incluyen:
- Talla normal o discretamente baja con proporciones corporales dentro de la normalidad.
- Articulaciones prominentes, habitualmente dolorosas, con movilidad restringida.
- Habitualmente cursan con cifosis torácica.
Los signos radiológicos más relevantes son los que siguen:
- Grados variables de aplanamiento de los cuerpos vertebrales.
- Irregularidades de las epífisis de los huesos largos.
El estudio radiológico no siempre ayuda a enfocar con precisión el diagnóstico.
⇒ Con estos signos clínicos y radiológicos hay que proceder al estudio molecular del gen COMP (19p13.1-p12), que codifica la proteína de la matriz oligomérica del cartílago (MIM 132400) (4). Si no se detectan mutaciones, será necesario analizar los siguientes genes: COL9A1 (MIM 120210), COL9A2 (MIM 600204), COL9A3 (MIM 600969) o MATN3 –(MIM 607078).
Es de interés señalar que los casos que cursan con afectación predominante de las rodillas más que de las caderas, son habitualmente debidos a mutaciones en COL9A1, COL9A2 o COL9A3.
3.1.2.4. Displasia epifisaria múltiple –autosómica recesiva-(MIM 226900)
También se conoce con el nombre de “Displasia epifisaria múltiple tipo 4”.
Los signos clínicos más relevantes, incluyen (12):
- Talla normal o discretamente baja.
- Dolor articular crónico.
- Deformidades moderadas de manos y/o pies.
Los signos radiológicos más relevantes son los que siguen:
- Epífisis aplanadas de las falanges.
- Aplanamiento de las epífisis femorales proximales.
- Rótula de doble capa.
⇒ Con estos signos clínicos y radiológicos hay que proceder al estudio molecular del gen DTDST. La mutación más frecuente es R279W en homocigosis. Cuando esta mutación se combina con otra más severa, se da lugar a una displasia diastrófica.
3.1.2.5. Discondrosteosis de Léri-Weill (MIM 127300) y displasia mesomélica de Langer (MIM 249700)
La presencia de mutaciones en el gen SHOX de los cromosomas sexuales en heterocigosis es responsable de un porcentaje elevado de casos de discondrosteosis de Léri-Weill, mientras que, en homocigosis, es responsable de la displasia mesomélica de Langer. La haploinsuficiencia de SHOX está presente en las pacientes con síndrome de Turner.
La discondrosteosis de Léri-Weill es una forma autosómica dominante de displasia mesomélica con deformidad de Madelung en el antebrazo (Figura 1).

Los signos clínicos más relevantes, incluyen (12):
- Deformidad de Madelung.
- Talla baja de tipo mesomélico.
- Hipertrofias muscular.
Los signos radiológicos más relevantes son los que siguen:
- Radio corto en relación al cúbito. Acortamiento y arqueamiento del radio con hipoplasia distal y luxación dorsal del cúbito.
- Huesos del carpo en cuña.
- Articulación radio-cúbito subluxada.
- Tibias cortas con prominencia de la porción medial de la tibia proximal.
⇒ Con estos signos clínicos y radiológicos hay que proceder al estudio molecular del gen SHOX (Xp22.3 e Yp11.3). Los casos en los que no se evidencia mutaciones en el gen SHOX pueden ser debidos a deleciones aguas abajo del SHOX que afecten elementos reguladores del mismo (13-15).
3.1.2.6. Displasia mesomélica de Langer (MIM 249700)
También denominada “discondrosteosis en homocigosis”, se transmite de forma autosómica recesiva en homocigosis dominante para el cuadro clínico de discondrosteosis.
Los signos clínicos más relevantes, incluyen (12):
- Talla baja por acortamiento de brazos.
- Micromelia mesomélica.
- Mandíbula hipoplásica.
Los signos radiológicos más relevantes son los que siguen:
- Acortamiento de huesos largos (Figura 2).
- Displasia distal del cúbito.
- Hipoplasia proximal del peroné.

⇒ Con estos signos clínicos y radiológicos hay que proceder al estudio del gen SHOX para el diagnóstico molecular.
3.1.3. Malnutrición
En los países en vías de desarrollo, la malnutrición junto con los procesos infecciosos y parasitarios, especialmente gastrointestinales, actúan de manera sinérgica en la génesis del fracaso de crecimiento; de hecho, el binomio infección-malnutrición (Figura 3), es con mucho la causa más frecuente de hipocrecimiento (1,2). En los países desarrollados, la escasez de alimentos es un situación excepcional y los hipocrecimientos de causa nutricional suelen ser secundarios a patologías crónicas, dietas inadecuadas en cantidad (ejercicio excesivo) o composición (vegetarianas estrictas o macrobióticas, entre otras) o trastornos más o menos graves de la conducta alimentaria (anorexia nerviosa, síndrome del miedo a la obesidad o fallo de medro, entre otros). El resultado final de todas estas situaciones es un aporte insuficiente de macro (malnutrición calórico-proteica) o de micronutrientes (minerales y vitaminas).

En la malnutrición calórico-proteica, una pérdida o inadecuada ganancia ponderal suele acompañar o preceder a la alteración del crecimiento en uno o dos años; si bien, no siempre es así, y el fracaso de crecimiento, acompañado de retraso de la EO y de la pubertad, puede ser el mecanismo de adaptación a la disminución de nutrientes, sin que se altere la relación peso/talla u otros marcadores clínicos/bioquímicos de malnutrición. En este proceso adaptativo, todo el sistema hormonal esta implicado (Figura 4), pero son de especial importancia, las alteraciones en el eje hipotálamo-hipófiso-gonadal (retraso puberal) y, especialmente, en el eje de la hormona de crecimiento (GH), donde se produce una insensibilidad a la GH con niveles séricos normales o aumentados de GH y disminuidos de IGF-I (factor de crecimiento semejante a la insulina número 1) e IGFBP-3 (proteína de transporte de IGFs número 3).
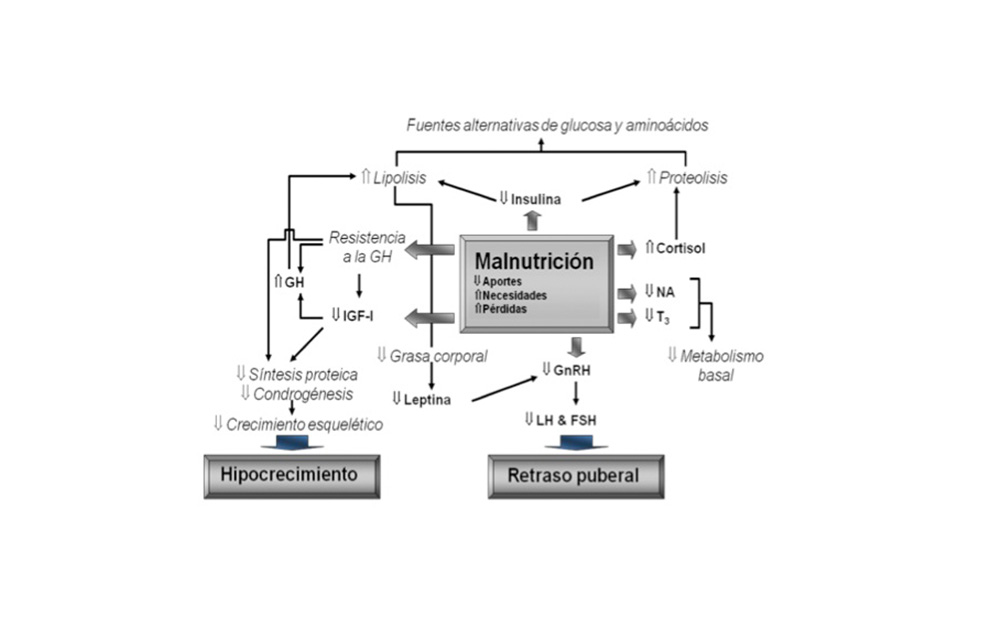
En lo referente a los déficits aislados de micronutrientes, se ha demostrado que, además de las deficiencias de calcio y fósforo, algunos oligoelementos como: iodo, hierro, cobre, cromo y zinc, son capaces de provocar cuadros de retraso de crecimiento en humanos. En los déficits vitamínicos aislados, la alteración del crecimiento no suele ser una manifestación clínica precoz, con excepción de la vitamina D, que al igual que el calcio y el fósforo, está directamente implicada en el proceso de mineralización y crecimiento del hueso.
3.1.4. Infecciones e infestaciones recurrentes
Son características de los países subdesarrollados, donde los procesos infecciosos y parasitarios, especialmente gastrointestinales, actúan de manera sinérgica con la malnutrición en la génesis del fracaso de crecimiento (Figura 3). En los países desarrollados, las infecciones recurrentes son poco frecuentes, pero cuando alteran el crecimiento, suelen reflejar la existencia de malformaciones anatómicas (renales y pulmonares, entre otras) o inmunodeficiencias subyacentes (SIDA) y contribuir al fracaso de crecimiento asociado a otras patologías crónicas. La malnutrición parece ser el principal mecanismo fisiopatológico implicado en estas formas de hipocrecimiento; ya que, la alimentación suplementaria durante los períodos de infección parece evitar o al menos disminuir sus repercusiones negativas sobre el crecimiento.
3.1.5. Enfermedades crónicas
El fracaso en el crecimiento y la talla baja consiguiente son hallazgos habituales en los niños que padecen enfermedades crónicas; de hecho, prácticamente, cualquiera de ellas puede producirlo si es lo suficientemente intensa y duradera (Tabla 1). Al menos, un 10-15 % de los hipocrecimientos es debido a enfermedades crónicas y es probable que se incremente a medida que lo haga la supervivencia de muchas enfermedades crónicas (1).
Los factores etiopatogénicos implicados en el fracaso del crecimiento asociado a las enfermedades crónicas son múltiples y varían dependiendo de la enfermedad de base, pero suelen estar en relación con: malnutrición, alteraciones metabólicas, efectos secundarios de la terapia, infecciones sobreañadidas y, posiblemente también, con los trastornos psicológicos que acompañan al padecimiento de una enfermedad crónica. Dentro de ellos, la malnutrición, a la que se puede llegar por múltiples mecanismos, es uno de los factores más importantes y constantes (1,2).
En lo que se refiere a las manifestaciones clínicas, las específicas de la enfermedad de base serán, en la mayoría de los casos, las predominantes en el cuadro clínico y el fracaso del crecimiento será sólo un síntoma más. No obstante, en determinadas patologías, como: el hipocrecimiento nutricional, la enfermedad celíaca, la enfermedad inflamatoria intestinal o la acidosis tubular renal, entre otras, el hipocrecimiento puede ser, durante años, el único o el principal síntoma de la enfermedad subyacente.
El patrón de crecimiento suele ser similar en todas ellas. La aparición de la enfermedad va a determinar un enlentecimiento más o menos marcado del ritmo de crecimiento que suele acompañarse de un retraso simultáneo de la maduración ósea y del desarrollo puberal. En caso de curación o mejoría significativa, se producirá una “crecimiento de recuperación” que puede permitir recuperar, total o parcialmente, la talla perdida. La medida en que el hipocrecimiento puede ser compensado depende de diferentes factores: edad de inicio, gravedad y duración del fracaso en el crecimiento, etiología y patogénesis de la enfermedad y, también, de factores individuales propios de cada sujeto.
3.1.6. Enfermedades endocrinológicas
3.1.6.1. Deficiencia de GH (DGH)
El porcentaje de retrasos de crecimiento de origen endocrinológico es muy escaso (5 %) y en concreto, las DGH suponen menos de un 1-2% de los hipocrecimientos (incidencia: 1:3.500-1:10.000 RN vivos). La DGH puede presentarse de forma aislada o asociado a otras deficiencias hipofisarias (hipopituitarismos) y puede ser congénita (alteraciones genéticas) o adquirida (tumores, traumatismos, histiocitosis, infecciones y radioterapia, entre otras). En la mayoría de los casos, la DGH es idiopática y sólo en aproximadamente un 20 % de los casos es posible identificar una causa orgánica responsable. En las formas idiopáticas, es frecuente el hallazgo en la RM craneal de determinadas anomalías morfológicas, como son: hipoplasia hipofisaria, tallo hipofisario ausente o muy reducido y neurohipófisis ectópica.
Estas anomalías estructurales, de etiopatogenia no aclarada pueden presentarse asociadas (síndrome de sección del tallo hipofisario) o separadas y su presencia conlleva un mayor riesgo de desarrollar otras deficiencias hipofisarias. Se supone que un 5-30 % de las formas idiopáticas tendrían una base genética, bien por mutaciones en el gen de GH (GH1; deficiencia o GH biológicamente inactiva) o del receptor de la hormona hipotalámica liberadora de GH (GHRHR), que condicionarían una DGH aislada (16), o bien por mutaciones en genes que codifican para factores de transcripción implicados en el desarrollo de la hipófisis (HESX1, POU1F1, PROP1, LHX3 y LHX4), que condicionarían deficiencias hipofisarias múltiples (Figura 5) (17,18).
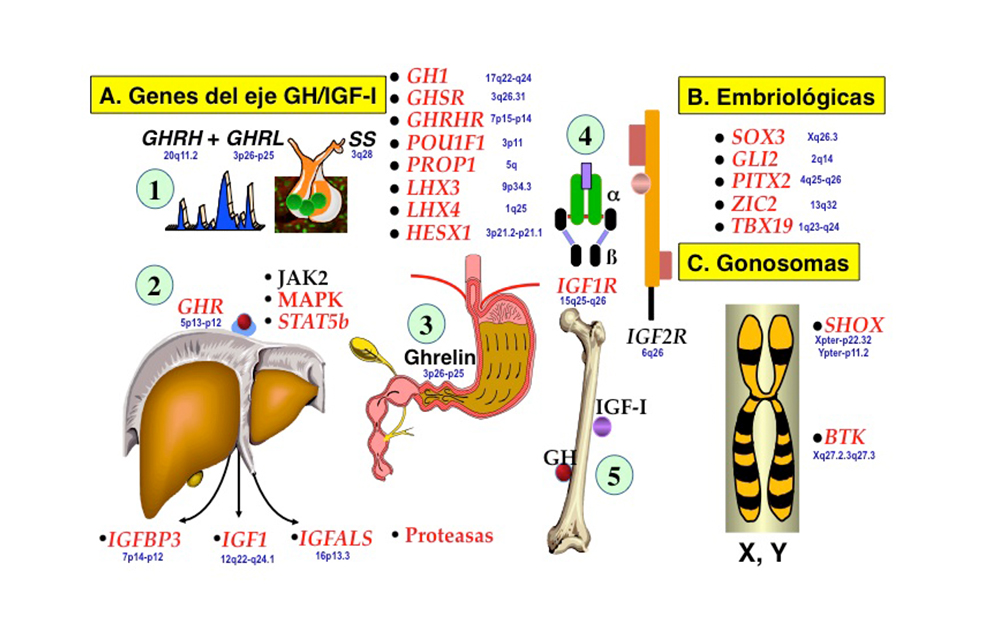
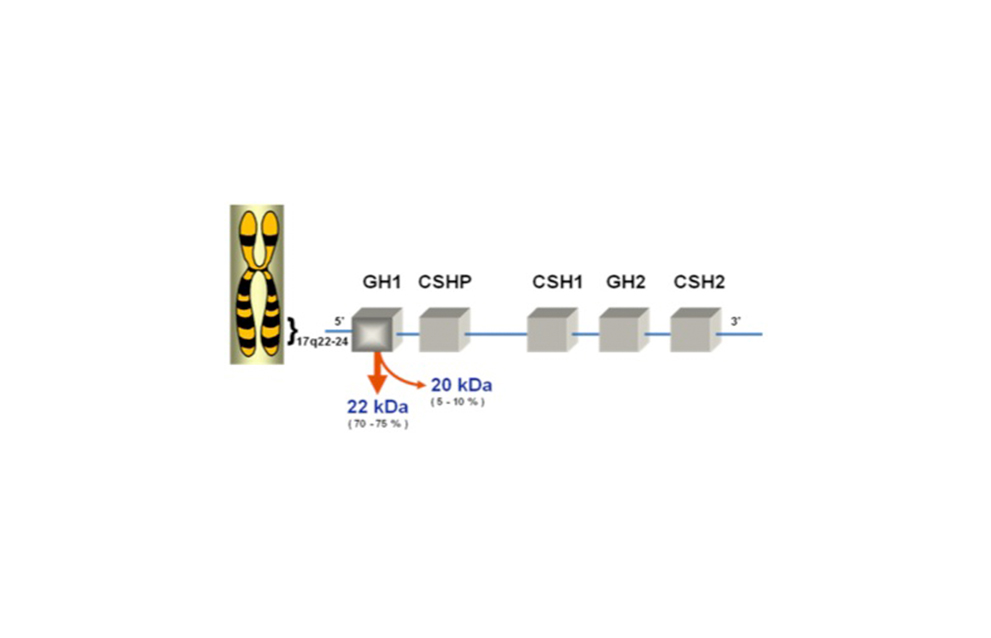
3.1.6.1.a Clasificación de la deficiencia aislada de GH
El gen de GH está conformado por 5 loci estructuralmente similares: GH1, CSHP, CSH1, GH2 y CSH2 en 17q22-24 (Figura 6).
El déficit genético de GH se asocia con al menos cuatro alteraciones mendelianas: 2 formas autosómicas recesivas (DAGH Tipo IA y IB), una forma autosómico dominante (DAGH Tipo II) y una forma ligada al cromosoma X (DAGH Tipo III).
DAGH Tipo IA
Fue descrita en 1970 por la Dra. Illig en tres niños suizos con alteración muy importante del crecimiento y aparente deficiencia de GH. La longitud al nacimiento es normal o ligeramente disminuida, con presencia de hipoglucemias agudas graves y retraso de crecimiento severo a los seis meses de vida postnatal (16). La respuesta al tratamiento con GH es extraordinaria, si bien en algunos casos la producción de anticuerpos contra GH puede detener el crecimiento. Pueden presentar mutaciones varias y deleciones en homocigosis de GH1 variables en tamaño (6,7kb, 7kb, 7,6kb y 45kb) (Figura 7).
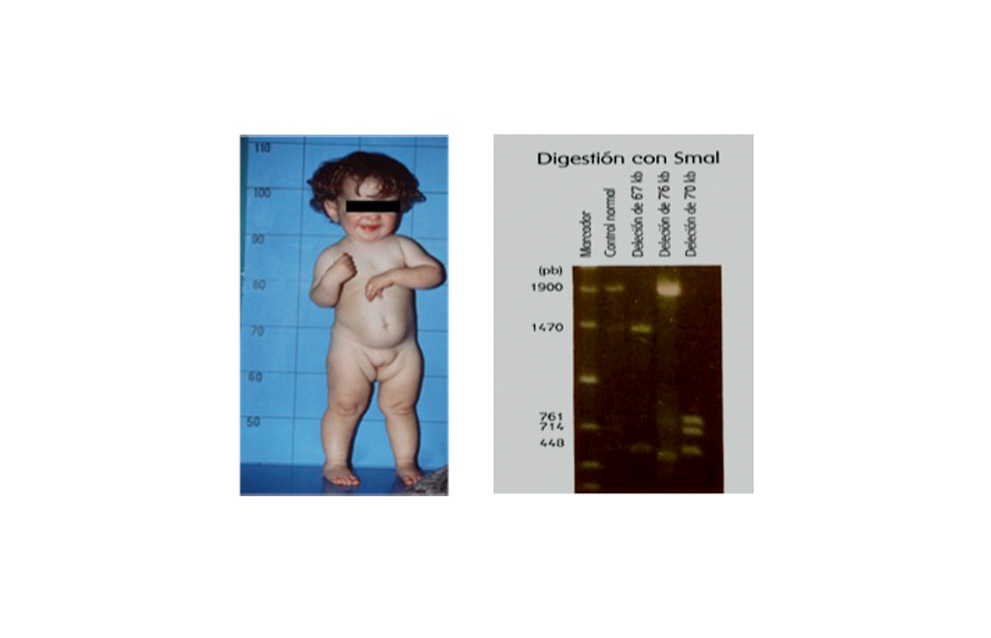
DAGH Tipo IB
Estos pacientes se caracterizan por presentar niveles bajos de GH, pero no indetectables, talla baja (<-2SDS para le edad y sexo) y velocidad de crecimiento <P25 para edad y sexo, con un marcado retraso de la edad ósea. Se transmite de forma mendeliana autosómica recesiva (dos padres no afectos, dos hijos afectos). El tratamiento con GH es muy favorable y no genera la producción de anticuerpos anti-GH. El fenotipo es variable y menos marcado por lo que a la deficiencia de GH hace referencia cuando se comparan con los pacientes con DAGH Tipo IA (2). Recientemente hemos diagnosticado una familia con mutación en GH1 (Figura 8) que está respondiendo de manera brillante al tratamiento con GH. Algunos pacientes presentan mutaciones en el gen GH1, mientras que otros, muestran mutaciones en el gen del receptor de GHRH (GHRHR).
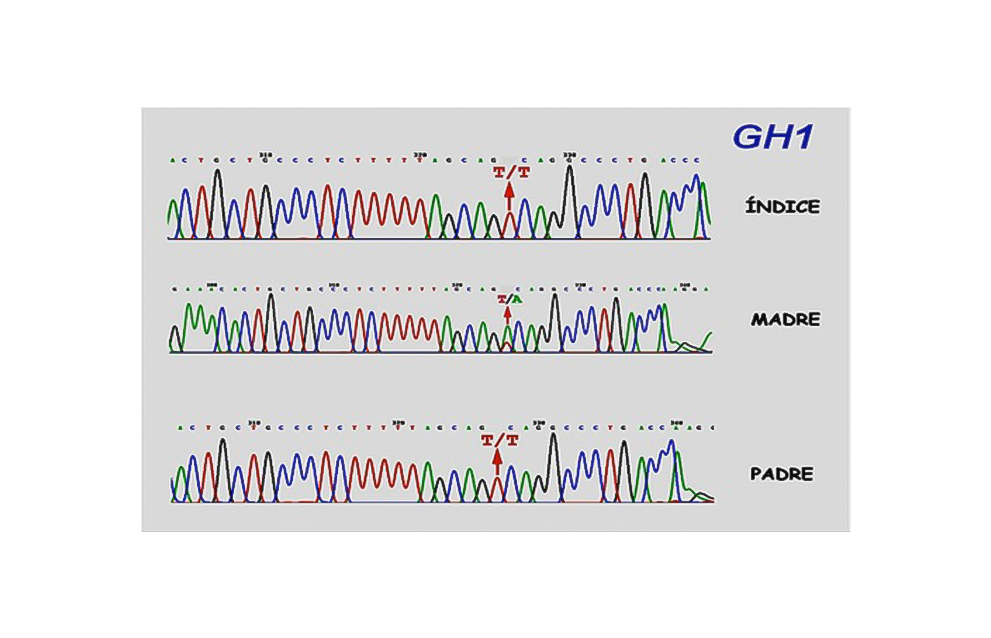
DAGH Tipo II
Se transmite de forma autosómica dominante. Es debida, fundamentalmente a mutaciones en el intrón 3 de GH1 (5’IVS-3). El fenotipo es similar al de los pacientes con deficiencia aislada de GH tipo IB. Es probable que la proteína de GH resultante sea de 17,5kd.
DAGH Tipo III
Se hereda de forma recesiva ligada al cromosoma X. Estudios recientes han demostrado que el brazo largo del cromosoma X se encuentra afectado por mutaciones y/o deleciones de una región que contiene dos genes, uno necesario para la producción normal de inmunoglobulinas y, el otro, para la expresión de GH. Por consiguiente, algunos de estos pacientes, además de la talla baja, presentan un cuadro clínico de agammaglobulinemia.
Hasta la fecha, no se han descrito mutaciones en el gen de GHRH ni en pacientes con DAGH Tipo IB ni en pacientes con DAGH Tipo II (2).
La manifestación clínica más característica de la DGH es el fracaso de crecimiento, que se acompaña de una marcada disminución de la velocidad de crecimiento y de retraso de la edad ósea. La secreción espontánea de GH y/o la respuesta de GH a los diferentes test de estimulación están disminuidas, al igual que los niveles séricos de IGF-I e IGFBP-3. En las formas congénitas o graves de inicio muy precoz, el hipocrecimiento puede estar presente ya en los primeros meses de vida y acompañarse de un fenotipo característico: cara de “muñeca”, voz aguda, incremento periabdominal de la grasa, acromicria, disminución de la masa muscular. Clásicamente, el DGH congénito se ha asociado a complicaciones perinatales (sufrimiento fetal, presentación podálica, fórceps, hipoglucemia e hiperbilirrubinemia conjugada), así como a un pene pequeño en los varones. En las formas adquiridas de inicio más tardío, el hipocrecimiento puede ser la única manifestación clínica.
3.1.6.1.b Deficiencia combinada de hormonas hipofisarias
La presencia de mutaciones en genes que codifican para factores de transcripción implicados en el desarrollo de la hipófisis (HESX1, SOX2, SOX3, POUIFI, PROP1, LHX3 y LHX4, POUIFI, PROP1) condicionan deficiencias hormonales hipofisarias combinadas. Las bases genéticas son complejas (17,18).
3.1.6.2. Insensibilidad a la GH (IGH)
Queda definida por la ausencia de una apropiada respuesta metabólica y de crecimiento a la GH endógena o a la GH administrada a dosis fisiológica de sustitución. La IGH adquirida es una situación clínica frecuente; ya que, se asocia a patologías crónicas y especialmente a la malnutrición calórico-proteica. Por el contrario, la formas de IGH congénitas son extraordinariamente infrecuentes (19,20). En la mayoría de los casos, se trata de mutaciones en homocigosis o heterocigosis compuesta en el gen del receptor de GH (GHR) (Figura 9) de herencia autosómica recesiva, que darían lugar a una insensibilidad total a la acción de la GH. El hipocrecimiento en estos casos es grave (síndrome de Laron) y el fenotipo característico, similar en muchos aspectos al de la deficiencia completa de GH.

Se han descrito también formas congénitas de insensibilidad parcial o completa a la GH debidas a: mutaciones en heterocigosis en el GHR, alteración de los mecanismos post-receptor encargados de la transmisión del mensaje de GH al interior de la célula (mutaciones en STAT-5b), deficiencias de la subunidad ácido lábil (mutaciones en IGFALS), deficiencias de IGF-I (mutaciones en IGF1) o resistencia a IGF-I (mutaciones en el gen del receptor de IGF-I, IGF1R), todas ellas de muy escasa incidencia.
Mutaciones en el gen de IGFALS
Hemos caracterizado clínica y molecularmente dos nuevas mutaciones en el gen de IGFALS que causan deficiencia completa de ALS en tres pacientes no relacionados con hipocrecimiento postnatal, niveles disminuidos de IGF-I e IGFBP-3, sin deficiencia de GH (21). Las curvas de crecimiento de los tres pacientes se reflejan en las gráficas representadas en la Figura 10.
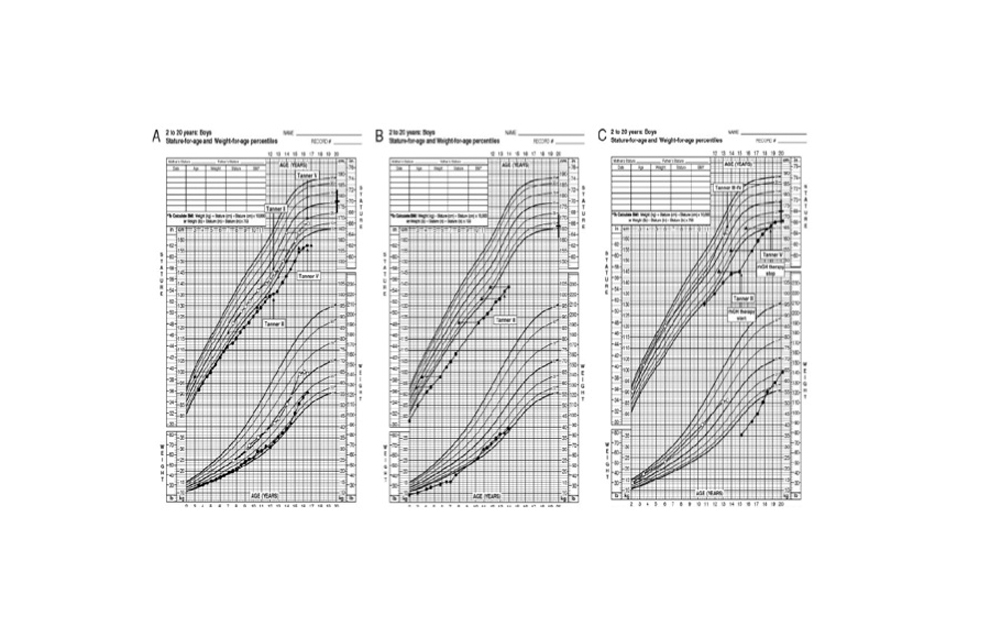
3.1.6.3. Nuevo mecanismo molecular responsable de deficiencia aislada de GH
El splicesosoma es un complejo de subunidades proteicas y de ARNsn que extrae los intrones de los pre-ARNm transcritos. El ARNhn contiene elementos de secuencias específicas que se reconocen y emplean durante el ensamblaje del splicesosoma.
En 2011 se describieron mutaciones recesivas en el ARNsn de U4atac (22,23), componente del splicesosoma menor, produciendo enanismo primordial microcefálico osteodisplásico tipo 1 (MOPD1), también conocido como síndrome de Taybi-Linder (TALS).
Recientemente hemos tenido ocasión de investigar una familia de padres no consanguíneos con cuatro hijas, tres de ellas con talla baja extrema postnatal –entre – 5 y y 6 SDS – que mostraban una deficiencia profunda de hormona de crecimiento, con niveles prácticamente indetectables para su edad y estadio puberal de IGF-I, IGFBP-3 y ALS. No existían evidencias de otras deficiencias hormonales asociadas (24). La resonancia magnética craneal puso en evidencia la existencia de hipoplasia hipofisaria en las tres hermanas afectas (Figura 11).

La respuesta al tratamiento con hormona de crecimiento ha sido excelente, incrementando más de 2 SDS de talla en el primer año de tratamiento.
Mediante análisis mutacionales y de segregación se descartaron anomalías en los genes del eje de GH: GH1, GHRHR, GHRH, POUIFI y PROP1. Por este motivo, se practicó un estudio del exoma completo que mostró la existencia de mutaciones bialélicas en el gen RNPC3 que codifica para la proteína de 65KD de U11/U12, una de los siete componentes proteicos en los di-snRNP de U11/U12, que media el procesamiento de los intrones tipo U12.
Dichas mutaciones se localizan en el segundo motivo de reconocimiento de ARN (RRM2) que se une al ARNsn U12 y es necesario para hacer un puente entre snRNPs de U11 y U12. Hemos observado una reducción estadísticamente significativa de los niveles celulares de los di-snRNPs de U11/U12, así como una regulación a la alza del ARNsn U4atac y di-ARNsn U4atac/U6atac, sugiriendo un posible mecanismo compensatorio.
Las investigaciones de ARNseq en muestras sanguíneas de los probandos mostró alteraciones del splicing de un conjunto de intrones U12, indicando la pérdida parcial de función consistente con los efectos patológicos moderados observados.
Por consiguiente, se trata de la primera familia descrita mundialmente con deficiencia aislada severa de hormona de crecimiento e hipoplasia hipofisaria y la segunda enfermedad humana causada por mutaciones en un componente específico del splisosoma dependiente de U12 por mutaciones bialélicas en el gen RNPC3, siendo la primera vez que se demuestra esta alteración en pacientes con deficiencia aislada de GH.
3.1.6.4. Descubrimiento de las primeras mutaciones en el gen de la proteasa PAPP-A2 y deficiencia de IGF-I libre
Los complejos trimoleculares de IGFBP-3 o IGFBP-5, IGF-I o IGF-II y ALS, de 150 kDa, ciculan en plasma con la imposibilidad de que IGF-I o IGF-II puedan ejercer sus acciones, hasta que dichos complejos son sometidos a las acciones de al menos dos proteasas, específicamente metaloproteinasas, PAPP-A y PAPP-A2, con actividad proteolítica específica de PAPP-A sobre IGFBP-4 y de PAPP-A2 sobre IGFBP-3 e IGFBP-5 (Figura 12).
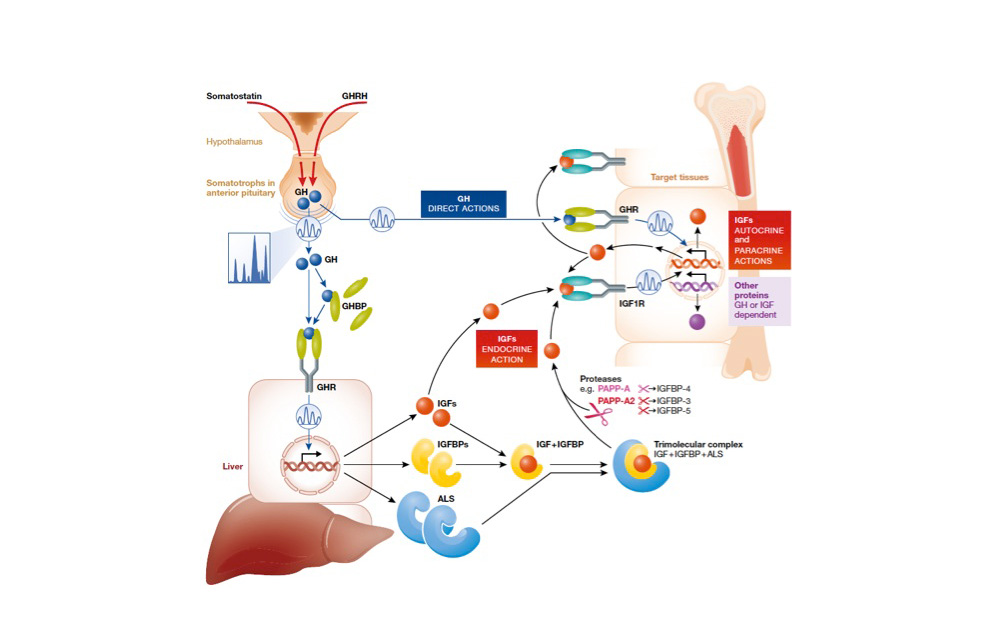
Mas recientemente se han descrito dos proteínas, estaniocalcinas 1 y 2 (STC1 y STC2) que, asimismo, intervienen en la regulación de la activación o inhibición de la señalización de IGF.
Recientemente hemos descrito las primeras mutaciones en PAPP-A2 (25) (Figura 13). La primera familia descrita, fue investigada y diagnosticada en el Servicio de Endocrinología del Hospital Infantil Universitario de Madrid (Figura 13, familia 1). Una niña y su hermano fueron referidos por presentar talla baja. En realidad, su talla se situaba dentro de la normalidad, pero muy alejada de su talla diana (al menos – 1,6 SDS por debajo de su talla diana o talla genética).
Los pacientes mostraban, en contraposición a lo esperado, niveles muy elevados en suero de IGF-I total, IGFBP-3, IGFBP-4, IGFBP-5 y ALS.
Para analizar las consecuencias de la función proteolítica de las mutaciones en el gen PAPP-A2 en nuestros pacientes, comparamos las variantes recombinantes con el gen normal de PAPP-A2, tras transfección de células HEK 293. El medio de las células transfectadas con ADNc que codifica para las mutaciones D643fs o A1033V, mostraron ausencia de actividad proteolítica para IGFBP-3 e IGFBP-5, mientras que las células con PAPP-A2 normal ejercía actividad proteolítica sobre estos sustratos.
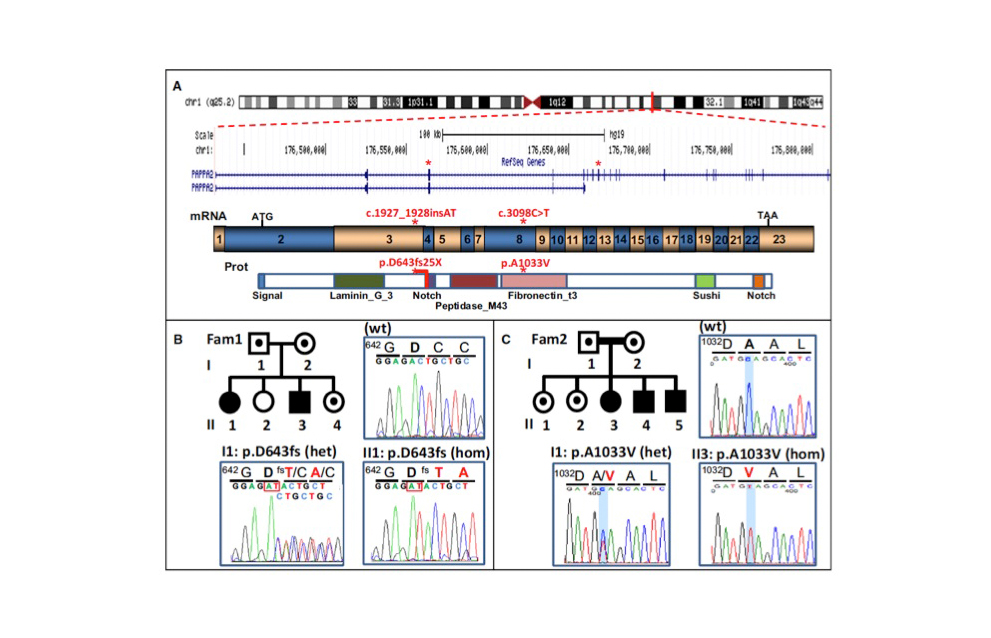
Estos pacientes han abierto una nueva vía a la comprensión de las funciones fisiopatológicas de IGF-I (26), al tiempo que hemos demostrado una buena respuesta en su crecimiento al tratamiento con IGF-I recombinante humano (27).
CONCLUSIONES
En conclusión, este manuscrito resume los fundamentos de las aportaciones efectuadas al diagnóstico de las bases moleculares de los hipocrecimientos, armónicos y disarmónicos, en donde el autor dirigiendo un amplio grupo de trabajo ha podido contribuir a diagnósticos complejos y novedosos en pacientes con deficiencia aislada de GH (tipos IA, IB y II), deficiencia combinada de hormonas hipofisarias, resistencia a la acción de la hormona de crecimiento, descubrimiento de nuevas mutaciones en el gen IGFALS y en la región PAR1 y gen SHOX en pacientes con discondrosteosis de Léri-Weill, displasia de Langer y talla baja idiopática, detallando, finalmente, las primeras mutaciones descritas mundialmente tanto en el gen RNPC3, asociadas con deficiencia familiar severa de GH e hipoplasia adenohipofisaria, como en el gen PAPP-A2 responsables de un nuevo síndrome asociado a talla baja postnatal, niveles elevados de IGF-I, IGFBP-3, IGFBP-4, IGFBP-5 y niveles indetectables de IGF-I libre.
AGRADECIMIENTOS
El autor desea expresar su sincero agradecimiento a todos los médicos, investigadores y técnicos, nacionales e internacionales, con quienes ha tenido la ocasión de colaborar activamente en la identificación de anomalías moleculares en pacientes con talla baja, ya armónica, ya disarmónica. Asimismo, desea expresar su agradecimiento a todos los pacientes y sus padres que han colaborado activamente en los estudios que se les han practicado. Finalmente, agradecimiento formal a cuantas fuentes de financiación han participado para la investigación desarrollada en este campo.
BIBLIOGRAFÍA
- Pozo J, Argente J. Hipocrecimiento armónico. En: Cruz-Hernández M, ed. Tratado de Pediatría M. Cruz (10ª edición). Madrid: Ergon S.A., 2011; págs: 916-931.
- Argente J, Pérez-Jurado LA. Genetic causes of proportionate short stature. Best Pract Res Clin Endocrinol Metab 2018; 32:499-522.
- Argente J. Challenges in the management of short stature. Horm Res Paediatr 2016;85:2-10.
- Rogol AD, Hayden GF: Etiologies and early diagnosis of short stature and growth failure in children and adolescents. J Pediatr 2014; 164(5 suppl):S1–S14.e6.
- Allen DB, Cuttler L. Short stature in childhood challenges and choices. N Engl J Med 2013; 368:1220–1228.
- Rosenfeld RG. Insulin-like growth factors and the basis of growth. N Engl J Med 2003; 349:2184-2186.
- Clayton PE, Cianfarani S, Czernichow P, Johannsson G, Rapaport R, Rogol A. Management of the child born small for gestational age through to adulthood: a consensus statement of the International Societies of Pediatric Endocrinology and the Growth Hormone Research Society. J Clin Endocrinol Metab 2007;92:804-810.
- Eggermann T, Begemann M, Binder G, Spengler S. Silver-Russell Syndrome: genetic basis and molecular genetic testing. Orphanet J Rare Dis 2010; 5:19.
- Azzi S, Salem J, Thibaud N, et al. A prospective study validating a clinical scoring system and demonstrating phenotypical-genotypical correlations in Silver-Russell syndrome. J Med Genet 2015; 52:446-453.
- Wakeling EL, Brioude F, Lokulo-Sodipe O, et al. Diagnosis and management of Silver-Russell Syndrome: first international consensus statement. Nat Rev Endocrinol 2017; 13:105-124.
- Ranke MB. Why treat girls with Turner syndrome with growth hormone? Growth and beyond. Pediatr Endocrinol Rev 2015; 12:356-365.
- Bonafe L, Cormier-Daire V, Hall C, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Genet A 2015; 167A(12):2869-2892.
- Benito-Sanz S, Thomas S, Huber C, et al. PAR1 deletions excluding the SHOX gene are found in Léri-Weill dyschondrosteosis (LWD) and idiopathic short stature (ISS). Am J Hum Genet 2005; 77:533-544.
- Benito-Sanz S, Gorbenko del Blanco D, Aza-Carmona M, et al. PAR1 deletions downstream of SHOX are the most frequent defect in a Spanish cohort of Léri-Weill dyschondrosteosis (LWD) probands. Hum Mut 2006; 10:1062-1062.
- Benito-Sanz S, Barroso E, Heine-Suñer D, et al. Clinical and molecular evaluation of SHOX/PAR1 duplications in Léri-Weill dyschondrosteosis (LWD) and idiopathic short stature (ISS). J Clin Endocrinol Metab 2011; 96: E404-E412.
- Phillips JA III, Cogan JD. Genetic basis of endocrine disease 6: molecular basis of familial human growth hormone deficiency. J Clin Endocr Metab 1994; 78: 11-16.
- Fang Q, George AS, Brinkmeier ML, et al. Genetics of Combined Pituitary Hormone Deficiency: Roadmap into the Genome Era. Endocr Rev 2016; 37: 636-675.
- Giordano M. Genetic causes of isolated and combined pituitary hormone deficiency. Best Pract Res Clin Endocrinol Metab 2016; 30: 679-691.
- Laron Z, Pertzelan A, Mannheimer S. Genetic pituitary dwarfism with high serum concentation of growth hormone–a new inborn error of metabolism? Isr J Med Sci 1966; 2:152-155.
- Godowski PJ, Leung DW, Meacham LR, et al. Characterization of the human growth hormone receptor gene and demonstration of a partial gene deletion in two patients with Laron type dwarfism. Proc Natl Acad Sci USA 1989; 86:8083-8087.
- Heath KE, Argente J, Barrios V, et al. Primary acid-labile subunit (ALS) deficiency due to recessive IGFALS mutations define a new form of postnatal growth deficit associated to low circulating IGF-I and IGFBP-3 levels, and hyperinsulinemia. J Clin Endocrinol Metab 2008; 93:1616-1624.
- He H, Liyanarachchi S, Akagi K, et al. Mutations in U4atac snRNA, a component of the minor spliceosome, in the developmental disorder MOPD I. Science 2001; 332: 238-240.
- Edery P, Marcaillou C, Sahbatou M, et al. Association of TALS developmental disorder with defect in minor splicing component U4atac snRNA. Science 2011; 332: 240-243.
- Argente J, Flores R, Gutiérrez-Arumí A, et al. Defective minor spliceosome mRNA processing results in isolated familial growth hormone deficiency. EMBO Mol Med 2014;6:299-306.
- Dauber A, Muñoz-Calvo MT, Barrios V, et al. Mutations in pregnancy-associated plasma protein A2 cause short stature due to low IGF-I availability. EMBO Mol Med 2016; 8:363-374.
- Argente J, Chowen JÁ, Pérez-Jurado LA, Frystyk J, Oxvig C. One level up: abnormal proteolytic regulation of IGF activity plays a role in human pathophysiology. EMBO Mol Med 2017; doi: 10.15252/emmm.201707950.
- Muñoz-Calvo MT, Barrios V, Pozo J, et al. Treatment with Recombinant Human Insulin-Like Growth Factor-1 Improves Growth in Patients With PAPP-A2 Deficiency. J Clin Endocrinol Metab 2016; 101: 3879–3883.
DECLARACIÓN DE TRANSPARENCIA
El autor/a de este artículo declara no tener ningún tipo de conflicto de intereses respecto a lo expuesto en la presente revisión.
ranm tv
Jesús Argente Oliver
Real Academia Nacional de Medicina de España
C/ Arrieta, 12 · 28013 Madrid
Tlf.: +34 91 159 47 34 | jesus.argente@uam.es
Año 2018 · número 135 (03) · páginas 242 a 253
Enviado*: 30.10.18
Revisado: 10.11.18
Aceptado: 12.12.18
* Fecha de lectura en la RANM