

Resumen
El linfoma difuso de célula grande B (LDCGB) constituye aproximadamente el 30% del total de los linfomas no Hodgkin (LNH). Es el más frecuente. En este artículo se analizan los factores pronósticos desde el punto de vista clínico, inmunohistoquímico y molecular, así como las estrategias terapéuticas actuales, incluyendo no solo el papel de la quimioinmunoterapia, sino también el del trasplante autólogo (autoTPH) y alogénico (aloTPH) de progenitores hematopoyéticos, las nuevas drogas y los resultados recientes con la inmunoterapia celular adoptiva, es decir, los avances clínicos que últimamente se han obtenido con la inmunoterapia con linfocitos T con receptores de antígenos quiméricos (LT RAQ).
Abstract
Diffuse large B-cell lymphoma (DLBCL) is the most common type of non Hodgkin lymphoma (NHL), accounting for about 30% of NHLs. This article discusses clinical, immunohistochemical and molecular prognostic factors, as well as current therapeutic strategies. The latter include chemoimmunoterapy, autologous and allogeneic transplantation of haematopoietic progenitors, new drugs and recent findings in celular adoptive immunotherapy. In other words, this article briefly examines the clinical advances that have recently been made with chimeric antigen receptor T Cell (CAR-T) therapy.
Palabras clave: LDCBG; Diagnóstico; Estrategia terapéutica; AloTPH; CART.
Keywords: DLBCL; Diagnostic; Therapeutic strategy; AloTPH; CART.
INTRODUCCIÓN
El LDCGB es el más común dentro del amplio grupo de LNH, suponiendo aproximadamente un 30% del total de esta variedad de linfomas. El segundo en orden de frecuencia, dentro de los LNH es el linfoma folicular y es menor la incidencia de otras variedades de LNH, tal como el linfoma linfocitico, el linfoma del manto y los linfomas de zona marginal entre otros tipos menos comunes. Los linfomas de origen T/NK forman aproximadamente el 12% del total de los LNH (1). En síntesis, más de la mitad de todos los LNH son la suma de LDCGB y linfoma folicular (Tabla 1).

El LDCGB es quimioinmunocurable en un 60-70% de los casos y representa un grupo heterogéneo de linfomas que son distintos desde el punto de vista clínico, en su origen celular y en los datos moleculares. Un 10-15% son refractarios al tratamiento de primera línea y otro 15-20% muestran recaída generalmente en los 2 ó 3 primeros años tras el tratamiento. Existen recaídas más tardías, aunque son poco frecuentes y en algunos casos quizás supongan la recidiva de un linfoma folicular transformado. La biopsia ganglionar es imprescindible para el diagnóstico, debiéndose practicar rutinariamente en la muestra los estudios inmunohistoquímicos y moleculares pertinentes.
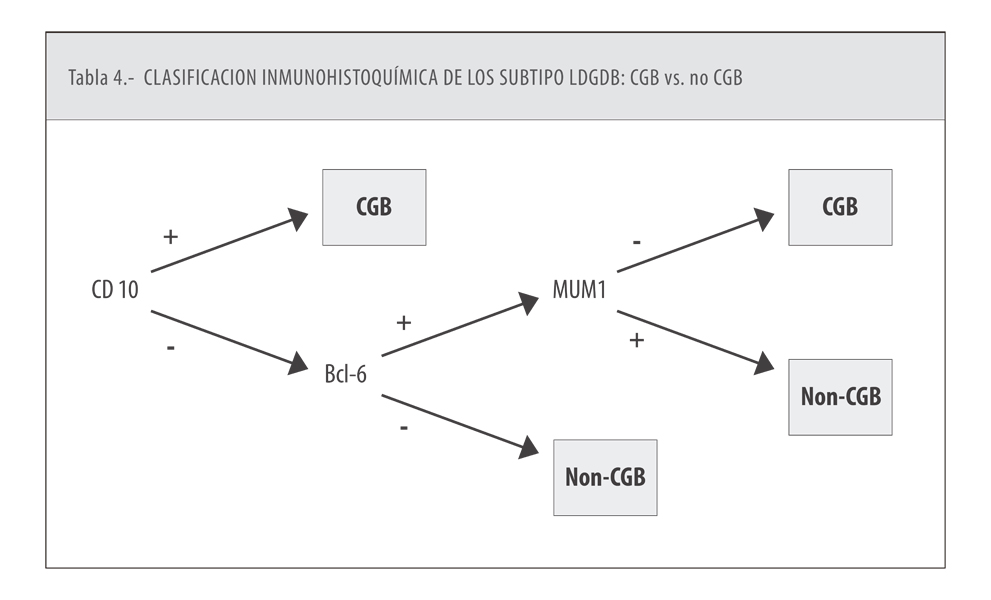
Se acepta internacionalmente desde hace 15-20 años que el LDCGB es un linfoma heterogéneo como veremos posteriormente, no solo desde el punto de vista clínico (2), sino también en cuanto a su origen celular (3,4) y características moleculares (Tabla 2). Desde el punto de vista del origen celular y teniendo en cuenta el perfil de expresión genética se distinguen 3 tipos: los de origen centro germinal (CG), los de célula activada (CA) y los inclasificables. Desde la práctica de la inmunohistoquímica y utilizando algunos algoritmos como el de Hans (5), se distinguen dos tipos: CG y no CG existiendo una alta correlación con los datos obtenidos por el perfil de expresión genética. Los estudios moleculares utilizando las técnicas de hibridación in situ (FISH) han mostrado que en un 5-10% de los LDCGB existe reordenamiento MYC con reordenamiento BCL-2 y/o BCL-6. Este tipo de linfomas se conoce con doble/triple hit (6). En ocasiones existe una sobreexpresión de proteínas MYC y BCL-2 y/o BCL-6 sin traslocación detectable por técnicas de FISH. Estos linfomas se conocen como doble expresadores (DE) (7).

PRESENTACIÓN CLÍNICA
Lo más habitual desde el punto de vista clínico es el debut con adenomegalias, bien localizadas, regionalizadas o diseminadas. Sin embargo, no es rara la participación extraganglionar. El curso de la enfermedad es muy agresivo y en caso de resistencia/recaída (RR) el número de remisiones completas obtenidas con quimioterapia de rescate es pequeño, cercano al 7% y la mediana de supervivencia de aproximadamente 6-7 meses.
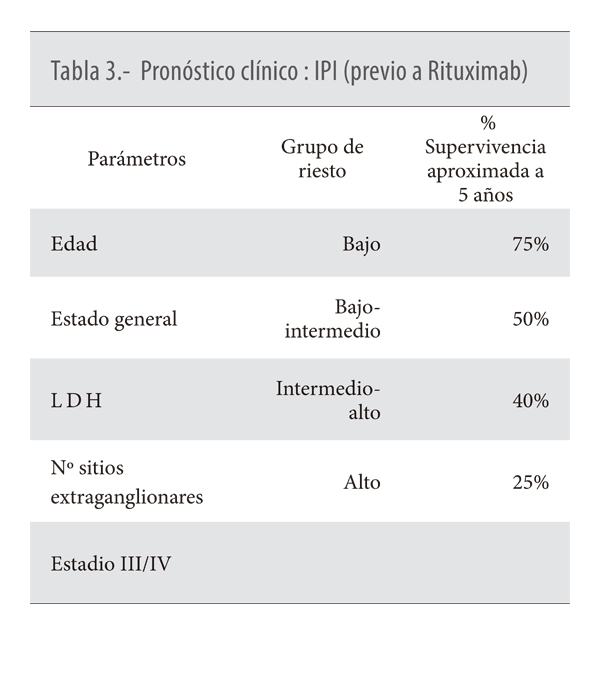
Se ha intentado establecer desde el punto de vista clínico el pronóstico de los LDCGB desde hace más de 25 años. Para ello se utilizaron una serie de datos clínicos, en el conocido como índice pronóstico internacional (IPI) (2). Estos datos son la edad, estado general, LDH, número de sitios con afectación extraganglionar y estadío clínico (Tabla 3). El IPI era capaz de discriminar el pronóstico de este tipo de linfomas, que oscilaba en lo que se refiere a la supervivencia global a los 5 años de unas cifras cercanas al 80% para los de bajo riesgo y unos números muy pobres alrededor del 25- 30% para los de riesgo alto (8). El valor del IPI no se modificó con la introducción de la inmunoterapia con Rituximab, el denominado R-IPI (2), que también era capaz de discriminar el pronóstico entre distintos grupos de enfermos.
HISTOLOGÍA, INMUNOHISTOQUÍMICA Y FISH
Debe de ser desestimada la práctica de la punción-aspiración con carácter diagnóstico. Es imprescindible la obtención de una biopsia adecuada del tumor. Es necesario limitar la biopsia con aguja gruesa a lesiones en zonas inaccesibles, que requieran para la obtención de la muestra cirugía abierta. Siempre se deben tener en cuenta las circunstancias clínicas del paciente. Sin embargo, hoy en día, existe una mayor tendencia por razones clínicas y logísticas al empleo de la biopsia con aguja gruesa, dado que no precluye la cirugía abierta posterior, si existe una duda diagnóstica razonable. El PET-TAC, actualmente rutina en la evaluación y el estadiaje del paciente con linfoma agresivo, puede señalar el lugar más interesante para biopsiar. El patrón histológico es característico, con una infiltración difusa por células grandes que destruyen la arquitectura ganglionar y que expresan antígenos pan-B, tal como CD19, CD20, CD22 y CD79a.
El estudio inmunohistoquímico es absolutamente necesario no solamente por razones diagnósticas sino para determinar el origen celular del tumor. El perfil de expresión genética no se utiliza en la práctica clínica por su alto coste y requerimiento del tejido fresco congelado. Empleando las técnicas inmunohistoquímicas y un algoritmo como el de Hans, ya hemos comentado que en razón al origen celular se distinguen 2 tipos: CG y no CG, que incluye CA, y los inclasificables, que suponen el 10-15% del total. Existe una buena correlación entre las conclusiones obtenidas a través del algoritmo de Hans con los datos del perfil genético. La concordancia es aproximadamente del 80%. Son de origen CG los CD1O+ o los que siendo negativos para el CDl0, son positivos para BCL-6 y negativos para MUM (Tabla 4). Existe un diferente pronóstico entre los LDCGB de origen CG y CA tras el tratamiento estándar de primera línea con el CHOP-R (9) (ciclofosfamida, adriamicina, vincristina, prednisona, y rituximab) obteniéndose claramente mejores resultados en los de origen CG.
El análisis por FISH del tejido ganglionar se ha convertido en rutinario, aunque no en todos los centros. Los LDCGB con reordenamiento MYC y BCL-2 y/o BCL-6 son clasificados como se ha señalado como linfomas doble o triple hit y suponen el 5-10% del total de estos linfomas agresivos, asociándose a un pronóstico claramente adverso. Cuando no existen las traslocaciones por FISH, pero si la sobreexpresión de estas proteínas en el estudio de inmuhistoquímica, se habla de linfomas doble expresadores DE que poseen un pronóstico intermedio entre los que se exhiben doble/triple reordenamiento y los que podríamos denominar convencionales. El estudio de las mutaciones no es habitual en la práctica médica entre otras razones por carecer de trascendencia clínica. Los linfomas con doble/triple reordenamiento han sido introducidos por el OMS en 2016 como una nueva categoría en la clasificación de los LNH, conociéndose como LNH de alto grado con reordenamiento c myc y BCL-2 y/o BCL-6 (7).
Los LDCGB con doble/triple reordenamiento en el 90% de los casos son de origen CG y expresan en el 95% de las ocasiones BCL-2. Muestran una elevada tasa proliferativa con Ki67 del 90%. Así mismo, muestran unas características clínicas adversas, presentándose en estadios avanzados con elevación de la LDH, afectación de médula ósea y otros lugares extraganglionares, y IPI alto y con un 15% de recaídas en el sistema nervioso central. El pronóstico es muy adverso con la quimioinmunoterapia de primera línea CHOP-R, con supervivencia global superior a los 2 años de solamente una cuarta parte de los enfermos. Es evidente la necesidad de una mejor terapia de primera línea para esta variedad de linfomas (10).
Los LDCGB DE no han sido catalogados por la OMS como una entidad clínica-patológica diferenciada. Los resultados terapéuticos obtenidos con la quimioinmunoterapia estándar de primera línea son intermedios entre los conseguidos con aquellos que muestran doble/triple reordenamiento y los que no lo muestran. Generalmente el origen de los linfomas DE es CA y es relativamente frecuente la afectación del sistema nervioso central. No existen estudios terapéuticos específicos para estos linfomas DE, pero es evidente el papel terapéutico insuficiente del CHOP-R. Se ha sugerido el uso inicial de quimioinmunoterapia tipo DA-EPOCH-R (dosis ajustadas de etopósido, prednisona, vincristina, ciclofosfamida, adriamicina y rituximab).
ESTRATEGIA TERAPÉUTICA ACTUAL DE LDCGB
Parece claro que a día de hoy, el tratamiento del LDCGB no puede ser uniforme, sino por el contrario debe atender a las diversas circunstancias clínicas, inmunohistoquímicas y moleculares de cada paciente.
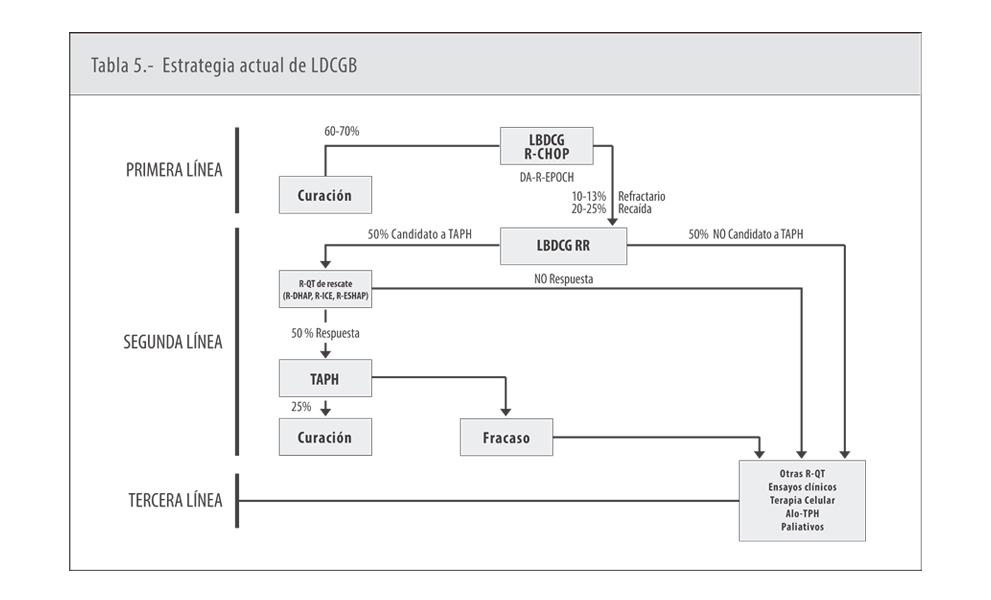
En primer lugar, comentaremos cual es el enfoque actual del tratamiento de los LDCGB sin doble/triple reordenamiento, ni DE. Debemos de distinguir los casos con afectación localizada y aquellos con afectación extensa. El 30-40% de los pacientes muestran afectación localizada y suelen ser de origen CG. En los estadios I no voluminosos con LDH normal, un tratamiento correcto son 3-4 ciclos de CHOP-R-21 y radioterapia en campo afecto (11). Para el resto de los estadios I y II es adecuado el uso de 6 ciclos de CHOP-R-21. En aquellos pacientes con afectación diseminada es correcto la terapia con 6-8 ciclos de CHOP-R-21. El CHOP-R-14 no supone ventaja terapéutica y por el contrario incrementa la toxicidad. Es posible la cura con los tratamientos referidos del 70% de los pacientes con LDCGB sin doble/triple reordenamiento y sin datos de DE.
Para los linfomas con doble/triple reordenamiento y aquellos DE, puede ser más útil la terapia con DA-EPOCH-R, más intensiva, aunque también más tóxica. En estas variedades de linfoma como ya se ha dicho los resultados terapéuticos son más pobres con la inmunoquimioterapia inicial.
En cualquier caso, el 10-15% de estos linfomas agresivos muestra refractariedad inicial al tratamiento y en un 20-25% de los casos sobreviene una recaída, generalmente en los 2-3 primeros años tras la terapia. En esta situación se debe de valorar al paciente para un eventual tratamiento con quimioinmunoterapia de rescate, seguida de autoTPH (12,13) en la estrategia actual de LDCGB. Si el paciente no es candidato a trasplante autólogo por diversas razones, se debe emplear quimioterapia de rescate, trasplante alogénico, ensayos clínicos si están disponibles, terapia celular o si la circunstancias del enfermo son muy precarias cuidados paliativos. Por el contrario, si el paciente es candidato al autoTPH se debe seleccionar una quimioinmunoterapia de rescate o de segunda línea, seguida del trasplante autólogo. No hay ventaja para el rescate de un tipo de quimioterapia sobre otra, tal como ESHAP-R (etopósido, cisplatino, arabinosido de citosina, prednisona y rituximab), DHAP-R (cisplatino, arabinosido de citosina, prednisona y rituximab), ICE-R (ifosfamida, carboplatino, etoposido) y otras. Nuestro grupo ha utilizado muy preferentemente el ESHAP-R, que permite una buena colecta celular y consigue resultados al menos comparables a los obtenidos con otras pautas. Es muy importante que exista quimiosensibilidad para proceder al trasplante. Con una experiencia acumulada de aproximadamente 25 años a nivel internacional, el 50% de los pacientes RR son susceptibles de quimioterapia de rescate y autólogo, y la mitad de los mismos, es decir aproximadamente el 25-30% del total de pacientes RR son largos supervivientes en remisión.
Si la terapia de segunda línea fracasa el paciente es candidato o bien a trasplante alogénico, alternativamente a terapia celular, ensayos clínicos, quimioterapias de tercera línea o cuidados paliativos.
En pacientes seleccionados con LDCGB se debe de realizar neuroprofilaxis (14). Son candidatos potenciales aquellos con un IPI alto, los pacientes con doble reordenamiento o DE y los que muestran algunas localizaciones específicas como afectación renal/suprarrenal, testicular, medula ósea, mama, ovario y otros. Desde el uso terapéutico del rituximab que penetra en el sistema nervioso central aunque de una manera relativamente escasa, ha disminuido la incidencia de meningiosis linfomatosa y la mayor parte de los pacientes con afectación del sistema nervioso central muestran infiltración parenquimatosa. No hay acuerdo en el tipo de neuroprofilaxis y el debate oscila entre la aplicación del metotrexate intratecal o sistémico a altas dosis. Quizá esto último sea lo más apropiado.
La estrategia terapéutica actual del LDCGB se resume en la (Tabla 5).
TRASPLANTE ALOGÉNICO DE PROGENITORES HEMATOPOYÉTICOS (TPHA)
Desde la década de 1980 el aloTPH primeramente de médula ósea y ulteriormente de progenitores de la sangre periférica han obtenido una masiva expansión internacional. En la actualidad el empleo del aloTPH es mucho más frecuente en procesos malignos mieloides (leucemia aguda mieloide y mielodisplasia) y el autoTPH en procesos linfoides, como linfomas y mielomas.
Sin embargo, existe una amplia experiencia con el alogénico en proceso linfoproliferativos y también concretamente en LDCGB. Usualmente, es una terapia utilizada tras fracaso del autólogo, es decir, en tercera línea en aquellos pacientes cuyas circunstancias lo permitan. En otras palabras, en pacientes que muestran quimiosensibilidad, ausencia de disfunción de órganos y una edad no muy avanzada. En buena parte el problema de la edad ha sido solventado con los nuevos procedimientos de aloTPH de intensidad reducida, útiles a cualquier edad especialmente en enfermos mayores y en aquellos que previamente han sufrido tratamientos intensivos de quimioterapia o autoTPH. Es conveniente un plazo de tiempo largo entre el autoTPH y el aloTPH para evitar toxicidad excesiva.
Datos recientes con un gran número de enfermos, concretamente con 1438 de aloTPH de intensidad reducida practicados entre 2008 y 2015 muestran similitud de supervivencia global y supervivencia libre de enfermedad con independencia del tipo de donante utilizado en LDCGB (15). Se emplearon donantes familiares HLA idénticos, donantes no emparentados con o sin depleción T y donantes haploidénticos utilizando ciclofosfamida postrasplante. Cerca del 40% de los pacientes mostraban supervivencia libre de progresión a los 3 años y la supervivencia global a 3 años fue aproximadamente un 50%. Claramente, el haplo de intensidad reducida con ciclofosfamida postrasplante, disminuyó la incidencia de enfermedad crónica injerto contra huésped.
Un estudio multicéntrico retrospectivo con 78 enfermos sometidos previamente a diversos tratamientos ha mostrado los resultados del aloTPH en linfoma doble hit y DE (16). La mitad de los casos mostraba refractariedad primaria y el 58% había sido sometido a autoTPH. Todos ellos eran pacientes jóvenes, sin comorbilidades y con enfermedad quimiosensible. Algo más de un 30% de los LDCGB con doble/triple reordenamiento mostraba una supervivencia libre de progresión a los 4 años.
NUEVOS FÁRMACOS
Numerosos fármacos han sido ensayados en el LDCGB tras fracaso de dos líneas de tratamiento. El bortezomib, la lenalidomida y el ibrutinib no han mostrado resultados claramente favorables y su uso fuera de ensayos clínicos es prácticamente inexistente. Se está investigando el papel de venetoclax y otras drogas. Voy a referirme brevemente a algunas de ellas.
El polatuzumab vedotina es un anticuerpo monoclonal conjugado anti CD79b con la monometil auristatina E (MMAE). El CD79b forma parte del receptor de la célula B y se expresa en la mayoría de estas, pero no en las células germinales. Este fármaco en LDCGB RR ha sido capaz de conseguir en monoterapia un 15% de remisiones completas y asociado a la Bendamustina un 39% de remisiones completas. Recientemente se ha comparado la combinación Polatuzumab vedotina-bendamustina-rituximab frente a bendamustina-rituximab en LDCGB RR.
Tanto la supervivencia global como la supervivencia libre de progresión con una mediana de seguimiento de 22 meses han sido claramente mejores en el primer grupo (17,18). Otros resultados interesantes han sido los obtenidos con MOR208 (anti CD19) combinada a lenalidomida. Así mismo, ha sido ensayado el anticuerpo biespecífico CD3/CD19 blinatumomab que consigue un 19% de respuestas tras 1 ciclo a costa de una toxicidad apreciable neurológica, hematológica y la necesidad de infusión continua durante 8 semanas (19).
INMUNOTERAPIA CELULAR CON LINFOCITOS T CON RECEPTORES ANTIGÉNICOS QUIMÉRICOS (LT RAQ o CAR T CELLS)
Como se ha citado previamente un 30-40% de los pacientes con LDCGB muestran resistencia primaria o recaída tras una remisión de duración variable. La mayoría de los enfermos sufren la recidiva en los primeros 2-3 años tras el tratamiento. La quimioinmunoterapia de rescate generalmente incluyendo platino, seguida de un autoTPH constituye la terapia estándar para todos aquellos pacientes que muestren clara quimiosensibilidad, en ausencia de comorbilidades. La práctica del autoTPH se ha extendido hasta los 70 años de edad, e incluso más en enfermos seleccionados. Sin embargo, solo la mitad de los pacientes con linfoma LDCGB en situación de RR son finalmente candidatos a esta segunda línea de tratamiento. Aproximadamente, un 50% no lo son por ausencia de quimiosensibilidad o padecimiento de comorbilidades. Del 50% de pacientes sometidos al procedimiento del autoTPH solo la mitad son largos supervivientes en remisión completa. En otras palabras, la segunda línea terapéutica consistente en la combinación de quimioinmunoterapia y autoTPH es capaz de curar al 25% de los hipotéticos candidatos a este procedimiento.
En síntesis, los pacientes no candidatos a autoTPH o los que recaen tras el mismo, son susceptibles de tratamiento con quimioterapia de tercera línea, nuevas drogas, ensayos clínicos, trasplante alogénico y recientemente la inmunoterapia con LT RAQ. En general, la quimioterapia como tercera línea ofrece unos resultados pobres, tanto en la consecución de remisiones completas, como en la supervivencia libre de enfermedad y la supervivencia global. Esta última se encuentra alrededor de 6-7 meses. El aloTPH es una opción en enfermos quimiosensibles y ofrece una probabilidad de curación aproximadamente a un tercio de los enfermos, a expensas de una alta mortalidad tóxica. Probablemente, estas cifras históricas pueden mejorarse con el empleo del aloTPH no mieloablativo o de intensidad reducida. El uso de ciclofosfamida postrasplante, fuera del contexto del trasplante alogénico haploidéntico, en mi opinión merece ser estudiado.
La reciente introducción de la inmunoterapia celular adoptiva con el empleo de LT RAQ ha modificado la panorámica terapéutica en los LDCGB (20). El tratamiento con LT RAQ, presenta algunas ventajas claras con respecto al aloTPH. No es relevante el factor edad y tampoco el factor quimiosensibilidad, este último clave su existencia para la indicación del aloTPH. También la inmunoterapia celular con LT RAQ posee otros inconvenientes. Entre ellos su alto coste, su complejidad técnica y logística y una toxicidad en absoluto desdeñable, singularmente el síndrome de liberación de citoquinas y la neurotoxicidad. La implantación de la terapia con LT RAQ ha sido enormemente facilitada por la comercialización de dos productos y posiblemente un tercero en breve plazo. Gilead (axicabtagene-ciloleucel) y Novartis (tisagenlecleucel) han logrado la comercialización de sus productos para el linfoma de célula grande B en RR a dos líneas de tratamiento y en el caso del último para la leucemia aguda linfoide infantil o juvenil multiresistente. Posiblemente el tercero Lisocabtagene-maraleucel (Celgene/Bristol) puede obtener aprobación en un futuro próximo.
El problema económico que supone este tipo de terapia es un desafio formidable para todos los servicios sanitarios de cualquier país, aunque probablemente lo atenúe el paso del tiempo, la práctica de un mayor número de procedimientos y eventuales soluciones imaginativas. Básicamente, el procedimiento utilizando la red científica y comercial de congelación y aislamiento de células T, inserción del receptor de antígeno quimérico con técnicas de ingeniería genética, expansión de los LT RAQ, congelación, y traslado para infusión intravenosa al paciente previa descongelación.
Para su práctica se requiere, con independencia de los mecanismos regulatorios de cada país, la acreditación por parte de las farmacéuticas implicadas.
Han sido publicados los datos de diversos ensayos, que consiguen aproximadamente un 50% de remisiones completas en casos de LDCGB RR persistiendo una alta proporción de las mismas tras un periodo de seguimiento que en algún estudio en concreto como el ZUMA 1 es de 27 meses. Este ensayo es el que ofrece un mayor tiempo de seguimiento (21). Una variedad de estudios clínicos se encuentran en marcha.
Entre las complicaciones tóxicas más comúnmente asociadas a la terapia con LT RAQ, primordialmente cabe citar el síndrome de liberación de citoquinas, la neurotoxicidad y la aplasia de células B. El síndrome de liberación de citoquinas es frecuente y precoz. El cuadro clínico se caracteriza por fiebre alta, hipoxia, taquicardia, hipotensión, disfunción cardiaca, edema pulmonar en el contexto de hipermeabilidad capilar, necesitando en los casos graves, ventilación mecánica. Puede observarse disfunción de otros órganos como hígado y riñón, citopenias o infección y ocasionalmente linfohistiocitosis hemafagocitíca. El síndrome de neurotoxicidad puede ser muy complejo, y ocurre en general algo más tardíamente con presencia de trastornos de la conciencia, alucinaciones, delirio, afasia, ataxia, déficit focales, somnolencia, y convulsiones. El edema cerebral puede ser muy grave (22).
Los factores que contribuyen al síndrome de liberación de citoquinas son básicamente la elevación de múltiples citoquinas producidas por las células infundidas y por otros linfocitos. La terapia consiste en la aplicación de tocilizumab un inhibidor del receptor IL-6 y esteroides. En general, la neurotoxicidad más seria se asocia a un síndrome de liberación de citoquinas más grave. No existe un acuerdo unánime sobre la clasificación de la severidad del síndrome de la liberación de citoquinas, habiendo sido propuestos varios sistemas. Estas diferencias en la gradación del síndrome de liberación de citoquinas pueden ser responsables, al menos en parte, de las diferencias de incidencia del mismo en los distintos ensayos clínicos.
La hipogammaglobulinemia consecuencia de la aplasia de células B, puede ser combatida con la administración periódica de inmunoglobulinas. Así como la morbilidad imputable al tratamiento con LT RAQ es notable, con frecuente necesidad de ingreso en la unidad de cuidados intensivos, la mortalidad referida es relativamente pequeña.
Independientemente de la toxicidad imputada a la terapia de LT RAQ, puede observarse la recaída del proceso linfomatoso. Estas recidivas han sido relacionadas con la pérdida de los LT RAQ y también con la desaparición del antígeno CD19 de las células tumorales.
Es previsible la extensión generalizada de la inmunoterapia con LT RAQ en el LDCGB RR, dado que los resultados son similares a los conseguidos con el aloTPH, tras la administración previa de dos líneas de tratamiento, y con menor toxicidad. Es cierto que aún no existe un seguimiento largo de los resultados con LT RAQ y no existen estudios comparativos directos y aleatorizados con el alogénico. Sin embargo, la terapia con LT RAQ se puede aplicar con independencia de la edad y quimiosensibilidad del linfoma, hechos que amputan muchas veces la posibilidad de trasplante. Se ha especulado con el papel puente de la terapia con LT RAQ para un aloTPH posterior. En cualquier caso, no deja de ser crítica la selección de pacientes para ejecutar este procedimiento. En la Tabla 6 se expresan algunos hechos y controversias entre el aloTPH y la terapia con LT-RAQ.

En Diciembre/19 en la sesión plenaria de la Asociación Americana de Hematología, se han comunicado resultados esperanzadores con un nuevo monoclonal biespecífico CD3 y CD20 en el tratamiento de los LNH. El Mosunetuzumab consigue remisiones completas en el LNH de mal pronóstico, incluyendo enfermos RR tras la terapia con LT RAQ. Estas remisiones con frecuencia parecen duraderas y el retratamiento es factible para pacientes que han alcanzado la remisión completa y posteriormente han recaído. ¿Potencial competidor para la terapia con LT RAQ?.
Sin ninguna duda, la experiencia clínica en años sucesivos, resolverá algunas de estas posibles controversias y es muy verosímil que podamos observar la amplificación de las indicaciones de la terapia con LT-RAQ, hoy limitada al LDCGB y leucemia aguda linfoblástica infantil-juvenil, a otras patologías.
BIBLIOGRAFÍA
- Armitage J, Weisenburger DD. New approach to classifying non Hodgkin’s lymphomas: clinical features of the major histologic subtypes. Non-Hodgkin’s Lymphoma Classification Project. J Clin Oncol. 1998;16(8):2780-2795.
- Sehn LH, Berry B, Chhanabhai M, et al. The revised International Prognostic Index (R-IPI) is a better predictor of outcome than the standard IPI for patients with diffuse large B-cell lymphoma treated with R-CHOP. Blood. 2007; 109(5):1857-1861.
- Rosenwald A, Wright G, Leroy K, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med. 2003;198(6):851-862.
- Sehn LH, Gascoyne RD. Diffuse large B-cell lymphoma: optimizing outcome in the context of clinical and biologic heterogeneity. Blood. 2015;125(1):22-32.
- Hans CP, Weisenburger DD, Greiner TC, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004; 103(1):275-282.
- Aukema SM, Siebert R, Schuuring E, et al. Double-hit B-cell lymphomas. Blood. 2011;117(8):2319-2331.
- Swerdlow SH, Campo E, Pileri SA, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375-2390.
- International Non-Hodgkin’s Lymphoma Prognostic Factors Project. A predictive model for aggressive non-Hodgkin’s lymphoma. N Eng J Med.1993;329(14):987-994.
- Mondello P, Mían M. Frontline treatment of diffuse large B-cell lymphoma: BeyondR-CHOP. Hematólogo Oncólogo 2019: 1-12.
- Cheah CYl, Oki Y, Westin JR, et al. A clinician’s guide to double hit lymphomas. Br J Haematol. 2015;168(6):784-795.
- Persky DO, Unger JM, Spier CM, et al. Phase II study of rituximab plus three cycles of CHOP and involved-field radiotherapy for patients with limited-stage aggressive B-cell lymphoma: Southwest Oncology Group study 0014. J Clin Oncol. 2008;26(14):2258-2263.
- Philip T, Guglielmi C, Ilagenbeek A, et al. Autologous bone marrow transplantation as compared with salvage chemotherapy in relapses of chemotherapy-sensitive non-Hodgkin’s lymphoma. N Engl J Med. 1995;333(23):1540-1545.
- Gisselbrecht C, Glass B, Mounier N, et al. Salvage regimens with autologous transplantation for relapsed large B-cell lymphoma in the rituximab era. J Clin Oncol. 2010;28(27):4184-4190.
- Schmitz N, Zeynalova S, Nickelsen M, et al. CNS International Prognostic Index: A Risk Model for CNS Relapse in Patients With Diffuse Large B-Cell Lymphoma Treated With R-CHOP. J Clin Oncol. 2016;34(26):3150-3156.
- Dreger P, Sureda A, Ahn KW, et al. PTCy-based haploidentical vs matched related or unrelated donor reduced-intensity conditioning transplant for DLBCL. Blood Adv. 2019;3(3):360-369.
- Herrera AF, Rodig SJ, Song JY, et al. Outcomes after Allogeneic Stem Cell Transplantation in Patients with Double-Hit and Double-Expressor Lymphoma. Biol Blood Marrow Transplant. 2018;24(3):514-520.
- Polson AG, Yu SF, Elkins K, et al. Antibody-drug conjugates targeted to CD79 for the treatment of non-Hodgkin lymphoma. Blood. 2007;110(2):616-623.
- Palanca-Wessels MC, Czuczman M, Salles G, et al. Safety and activity of the anti-CD79B antibody-drug conjugate polatuzumab vedotin in relapsed or refractory B-cell non-Hodgkin lymphoma and chronic lymphocytic leukaemia: a phase 1 study. Lancet Oncol. 2015;16(6):704-715.
- Cohen JB. Novel therapies for relapsed/refractory aggress1ve lymphomas. Hematology Am Soc Hematol Educ Program. 2018(1):75-82.
- Chavez JC, Bachmeier C, Kharfan-Dabaja MA. CAR T-cell therapy for B-cell lymphomas: clinical trial results of available products. Ther Adv Hematol 2019;10:1-20.
- Locke FL, Ghobadi A, Jacobson CA et al. Long-term safety and activity of axicabtagene ciloleucel in refractory large B-cell lymphoma (ZUMA-1): a single-arm, multicentre, phase 1-2 trial. Lancet Oncol. 2019;20:3 l-42.
- Brudno JN, Kochenderfer JN. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019;34:45-55.
DECLARACIÓN DE TRANSPARENCIA
El autor/a de este artículo declara no tener ningún tipo de conflicto de intereses respecto a lo expuesto en la presente revisión.
ranm tv
José María Fernández-Rañada de la Gándara
Real Academia Nacional de Medicina de España
C/ Arrieta, 12 · 28013 Madrid
Tlf.: +34 91 159 47 34 | E-Mail: jmranada@yahoo.es
Año 2020 · número 137 (01) · páginas 27 a 34
Enviado*: 18.02.20
Revisado: 24.02.20
Aceptado: 20.03.20
* Fecha de lectura en la RANM