

Resumen
La LHH es una enfermedad concreta y siempre un síndrome caracterizado por una excesiva activación inmune y datos clínicos y analíticos de hiperinflamación, que conduce a una supervivencia corta si se demora o no se aplica el tratamiento adecuado. Ha sido una entidad poco diagnosticada, cuya incidencia aparentemente ha aumentado en época reciente, quizá debido a su mayor reconocimiento. Los casos de base genética/familiar ocurren en la infancia por un trastorno heredado de una función citotóxica deficiente de las células T/NK. En el adulto la mayor parte de los pacientes disponen de causas predisponentes, tal como infección singularmente por virus Epstein-Barr, neoplasias más comúnmente por linfomas no Hodgkin de origen T, procesos autoinmunes y rara vez algunos pacientes son catalogados en este sentido como idiopáticos. El tratamiento se basa en la administración de la pauta LHH-94, propiciada por la Sociedad Histiocítica, requiriendo todos los casos de base genética la aplicación ulterior de un trasplante alogénico de progenitores hematopoyéticos , lo que también es una opción terapéutica para los adultos con enfermedad resistente, progresiva o recidivante.
Abstract
HLH is a specific disease and always a syndrome characterized by excessive immune activation and clinical and analytical data of hyperinflammation that leads to a short survival if the appropriate treatment is delayed or not applied. It has been a poorly diagnosed and its incidence has apparently increased in recent times, perhaps due to its greater recognition. Genetic /familial-based cases occur in childhood from an inherited disorder of a poor cytotoxic function of T / NK cells. In adults, the majority of patients have predisposing causes, such as Epstein Barr virus infection, neoplasms most commonly due to non-Hodgkin lymphoma of T origin, autoimmune processes, and rarely are some patients classified as idiopathic. Therapy is based on the administration of the LHH-94 treatment protocol, promoted by the Histiocytic Society. All cases with a genetic base require the subsequent application of an allogeneic transplantation of hematopoietic progenitors, which is also a therapeutic option for adults with disease resistant, progressive or recurrent.
Palabras clave: Hemafagocitosis; Etopósido; Linfomas T periféricos.
Keywords: Hemophagocytosis; Etoposide; Peripheral T-cell lymphoma.
INTRODUCCIÓN
La linfohisticitosis hemofagocítica (LHH) es un trastorno raro y con frecuencia fatal, imputable a una marcada actividad inmune que conduce a una situación generalizada de hiperinflamación, tanto desde el punto de vista clínico como analítico y finalmente a un fracaso multiorgánico. La enfermedad puede observarse a cualquier edad aunque más comúnmente en niños menores de 1 año, puede ocurrir con carácter familiar o esporádico y pueden existir o no factores estimulantes, singularmente la infección, tanto en casos con predisposición genética como en pacientes esporádicos. En los últimos tiempos ha sido descrita un aumento en la incidencia de la LHH, cuya causa es oscura y quizá pueda deberse a un incremento en el conocimiento de esta entidad y por tanto un mayor número de diagnósticos.
Tradicionalmente, los casos han sido clasificados como primarios o bien de carácter secundario (1). Las LHH primaria se debe a mutaciones genéticas que dificultan la función citotóxica de las células T y NK. Típicamente estos casos aparecen en la primera infancia y tienen con frecuencia carácter familiar. El modelo de herencia es autosómico recesivo. Múltiples mutaciones han sido involucradas en la etiología de esta afección, desde la identificación del gen de la perforina hace aproximadamente 20 años (2). Una variada sinonimia ha sido paralela al progresivo conocimiento de este síndrome desde su descripción original en 1934 (3). Estos problemas terminológicos han traducido sin duda el grado de confusión que ha existido durante mucho tiempo acerca de este proceso.
En la opinión de algunos autores más que de pacientes con LHH primaria o secundaria, debía de hablarse de enfermedad LHH para referirnos a pacientes con predisposición genética, identificándose mutaciones que afectan a la función citotóxica de las células CD8+ y NK y a otros enfermos con una asociación etiológica concreta como infección, enfermedad neoplásica, trastornos autoinmunes y algunos pacientes que padecen LHH con carácter idiopático. Por otro lado, existiría enfermedad LHH símil, muy bien ejemplificada por el síndrome de excesiva activación inmune e hiperinflamación que puede ocurrir en la sepsis y en otras circunstancias clínicas (4).
DIAGNÓSTICO DE LA LHH. ASPECTOS GENERALES
Aunque no es el propósito de este artículo la descripción de la etiología, el diagnóstico y el tratamiento de la LHH en la infancia, se debe recalcar que muchos de nuestros conocimientos en el adulto provienen de lo aprendido en niños con esta afección (5,6).
Así pues como se observa en la Tabla 1 los criterios expuestos para el diagnóstico de la LHH fueron elaborados en pacientes pediátricos por la Sociedad Histiocítica en el 2004 y han sido también aplicados para la catalogación diagnóstica de pacientes adultos.

Lo mismo puede decirse con respecto a los avances terapéuticos, también basados en un protocolo de la Sociedad Histiocítica desarrollado en 1994 y modificado en el 2004 combinando etopósido y dexametasona en una pauta de 8 semanas de duración seguidas tardía o inicialmente por la administración de ciclosporina A. Todos los casos de LHH de base genética son mortales no tratados y aunque el tratamiento basado en etopósido/dexametasona mejora las perspectivas de respuesta y supervivencia ni un solo caso ha sido curado sin la práctica de trasplante alogénico de progenitores hemopoyéticos (7,8,9).
En el adulto, la mayoría de los pacientes con LHH son enfermedades con una etiología bien definida y básicamente asociados a factores predisponentes tal como infección, preferentemente por virus Epstein-Barr, neoplasia, comúnmente por linfoma no Hodgkin (LNH), singularmente de origen T/NK, trastornos reumatológicos como la artritis reumatoide juvenil, el lupus eritematoso diseminado, vasculitis, dermatomiositis, inmunodeficiencia o trastornos inflamatorios intestinales. En los últimos años, la LHH asociada a los trastornos autoinmunes citados, ha sido denominado síndrome de activación de los macrófagos (SAM) y esta entidad tiene unas connotaciones terapéuticas diferentes, omitiéndose la aplicación de etopósido. En algunos casos no es posible discernir un factor etiológico y estos pacientes son calificados como LHH de carácter idiopático.
Los factores predisponentes y provocadores de la excesiva activación inmune tanto en niños como en adultos que inducen a un empeoramiento de la función citotóxico de las células T y NK son frecuentemente infecciones bien virales, bacterianas, fúngicas o protozoarias. Entre las infecciones virales cabe destacar por su incidencia el virus Epstein-Barr, citomegalovirus, otros virus herpes, el HIV y el parvovirus. Entre otros microorganismos implicados cabe citar las micobacterias y la leishmaniosis (10).
DIAGNÓSTICO DE LA LHH EN EL ADULTO
Es esencial como en los casos pediátricos, un diagnóstico precoz. El diagnóstico es fundamentalmente un ejercicio clínico, y se basa en un conjunto de síntomas, signos, y datos de laboratorio, no existiendo ni un solo elemento patognomónico. Es un apoyo importante para el diagnóstico en el adulto los criterios emitidos por la Sociedad Histiocítica en el 2004 para los enfermos pediátricos. Se exige la presencia de al menos 5 de los 8 criterios emitidos, aunque no siempre estos están presentes al principio de la evolución. Ante un alto índice de sospecha, es totalmente recomendable el inicio del tratamiento dada la letalidad de la afección y el hecho bien reconocido de la posible ausencia de varios de los posibles criterios en la fase temprana de la LHH. Los 8 criterios citados son: fiebre, esplenomegalia, citopenias, hipertrigliceridemia y/o hipofibrinogenemia, hemofagocitosis, hiperferritinemia, trastornos de la función NK y niveles altos de CD25s. Existen otros muchos datos clínicos y analíticos adicionales, como hepatomegalia, transaminitis, coagulopatía, hipoalbuminemia, edema, rash, hiponatremia, y elevación del dímero-D. Es frecuente la participación del sistema nervioso central con trastornos muy variables, tal como signos focales, convulsiones, ataxia, disartria y encefalopatía (11). La gran mayoría de pacientes con LHH y con sintomatología neurológica también tienen síntomas sistémicos, aunque la participación del sistema nervioso central en algunos pacientes es la única alteración demostrable. Las reactivaciones en el sistema nervioso central pueden ocurrir en el curso evolutivo de la LHH con independencia o no de las manifestaciones sistémicas. El líquido cefaloraquideo (LCR) muestra alteraciones como pleocitosis e hiperproteinoraquia. Existen casos atípicos como aquellos que pueden evolucionar con fallo hepático agudo único o acompañado por otras manifestaciones de hiperinflamación.
No existe una guía aceptada con carácter universal para el diagnóstico de la LHH en el adulto y básicamente su reconocimiento implica destreza clínica en la valoración de un complejo cuadro clínico-biológico. Conviene ser meticuloso en el análisis clínico para descartar las situaciones LHH símiles especialmente la sepsis y algunas hemopatías malignas como LNH y leucemia aguda. Se debe de ser agresivo en la metodología diagnostica para identificar los pacientes con septicemia y linfoma/leucemia practicando los estudios microbiológicos y las biopsias de médula ósea o ganglionares oportunas antes de comenzar un tratamiento que incluya esteroides y que puede desfigurar el cuadro clínico y el origen de la enfermedad. Además de las presentaciones sepsis-símiles hay que tener muy presente los cuadros de infección de otra naturaleza, el síndrome de activación de los macrófagos y las subyacente a etiología autoinmune, así como la LHH que se observa en el paciente inmunocomprometido tal como ocurre con los enfermos tratados con azatioprina y mercaptopurina por enfermedad inflamatoria intestinal y también los casos de LHH que modernamente se observan tras la aplicación de nuevas formas de inmunoterapia celular y anticuerpos biespecificos que dan lugar a síndromes de hiperinflamación que exigen tratamiento.
Es necesario realizar algunos comentarios acerca de diversos parámetros diagnósticos. En primer lugar, resaltar que no hay ningún dato específico, que la sensibilidad de algunos parámetros es claramente variable y que no se debe demorar el tratamiento ante una sospecha clínica fundada, aun no observándose los 5 criterios mínimos exigidos por la Sociedad Histiocítica en el 2004 (12).
La hemofagocitosis es habitualmente detectada en la medula ósea y eventualmente en otros tejidos, como el ganglio linfático. Puede faltar en etapas tempranas en la LHH y en absoluto excluye el diagnostico. Por otro lado, la hemofagocitosis no es específica y puede observarse con cierta frecuencia en pacientes gravemente enfermos por distintas dolencias, tal como la sepsis.
Tampoco es específica la hiperferritinemia aunque los niveles muy altos por encima de 10.000 ng/ml son sugestivos de la LHH . No hay que olvidar que la ferritina se comporta como un reactante de fase aguda y puede estar elevada en casos de infección, inflamación, tumor y en pacientes con fallo hepático debido a necrosis de los hepatocitos con suelta de ferritina.
Un nivel normal de CD25s, prácticamente excluye el diagnostico de LHH aunque no es un parámetro especifico, ya que además de ser un marcador de la activación de las células T, esta también asociado a la proliferación presente en diversas neoplasias (13). Otros marcadores biológicos útiles para el diagnóstico son los niveles CXCL9 indicativo de la actividad del interferón-gamma o la IL18 sugestiva de actividad inflamatoria. El nivel de CD163s es un marcador de activación de los macrófagos que se observa elevado en la artritis reumatoide juvenil u otros trastornos autoinmunes. Desde el punto de vista etiológico de la LHH del adulto el esfuerzo diagnostico debe de ser máximo ya que tiene connotaciones terapéuticas. En la Tabla 2 se presenta un esquema de enfoque diagnóstico.
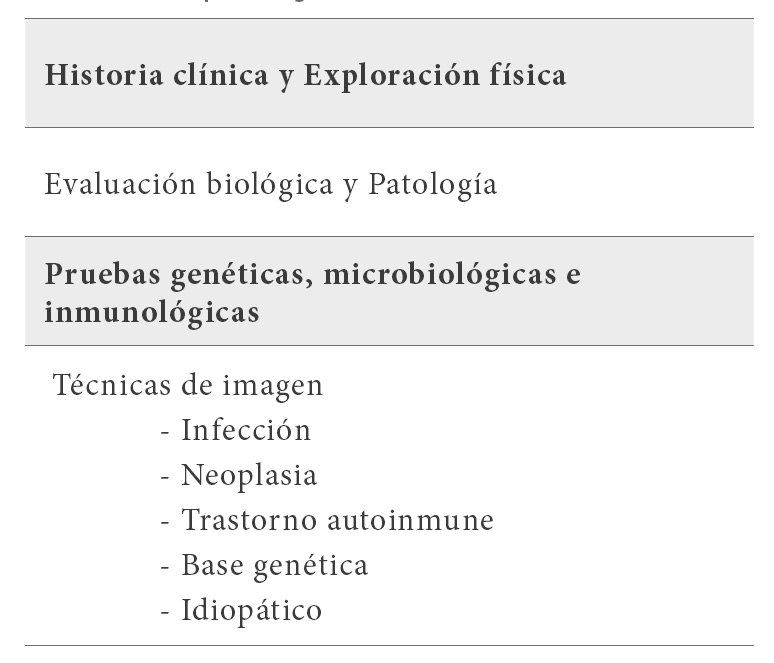
A continuación exponemos los casos observados de LHH en el Hospital Quirón Madrid de LHH en los últimos años.
CASO 1.-
Mujer de 38 años de edad que a las 21 semanas de su segundo embarazo presentó un cuadro de fiebre alta, malestar, artralgias, mialgias, pancitopenia, hepatoesplenomegalia, transaminitis, aumento de los reactantes de fase aguda e infiltrados intersticiales pulmonares bilaterales, ingresando el 13/Enero/20 siendo tratada con Ceftriaxona y Azitromicina. A las 48 horas mostró vómitos, diarreas, dolor abdominal, tendencia a la hipotensión, metrorragias, insuficiencia renal y progresión de la pancitopenia por lo que fue ingresada en UCI. Al día siguiente, tras administración de oxitocina, expulsa un feto muerto. Todos los estudios microbiológicos realizados fueron negativos exceptuando positividad de antígeno temprano para el virus Epstein-Barr. La evolución en UCI mostró rápida progresión de los infiltrados pulmonares y fracaso multiorgánico, incluyendo importante elevación de las transaminasas, coagulopatía, trombopenia severa y fracaso renal agudo. Precisó intubación y administración de noradrenalina. Los estudios de imagen evidenciaron importante derrame pericárdico, que requirió pericardiocentesis, derrame pleural bilateral y abundante líquido ascítico.
Ante la posibilidad, de un síndrome hemofagocítico, se practicaron estudios adicionales: el aspirado de médula ósea no mostró hallazgos relevantes, la biopsia de médula ósea documentó un síndrome hemofagocítico asociado a eritroblastopenia y el estudio inmunológico de los linfocitos en sangre periférica se objetivó un aumento de linfoticos T activados, disminución de linfocitos T citotóxicos, sin alteración en la expresión de las perforinas en las células NK. Otros datos fueron hiperbilirrubinemia, hiperferritinemia que llegó a 22.000 ng/ml e hipofibrinogenemia. Fue tratado según el protocolo LHH 2004 con dexametasona y etopósido, con profilaxis antiinfecciosa con fluconazol, aciclovir y septrim forte. La terapia obtuvo tanto mejoría clínica como analítica, a pesar de neutropenia severa que requirió CSF-G. En este momento la PCR cuantitativa para Epstein-Barr fue positiva por lo que se asoció al tratamiento Rituximab e inmunoglobulinas iv. La paciente continuó con su mejoría y pocos días después el DNA virus Epstein-Barr era negativo. Se extubó a la paciente. Sin embargo, unos días más tarde mostró hemorragia vaginal importante y progresiva que obligó a trasfusión repetida y finalmente a histerectomía. Las complicaciones observadas fueron importante miopatía, necrosis distal por aminas administradas en la UCI a consecuencia del shock refractario y desnutrición.
Dado que la paciente padece una sordera sensorial de 6 años de evolución se realizó un estudio molecular de las mutaciones en probable relación con síndrome hemofagocítico sobre todo la STXBP2 que se asocia con sordera. Todos los resultados genéticos fueron negativos.
Con el diagnóstico de síndrome hemofagocítico en probable relación con infección VEB la enferma fue dada de alta en buen estado.
CASO 2.-
Mujer de 23 años que comienza con faringitis, náuseas, ictericia progresiva y coluria el 28/Febrero/2020 por lo que fue ingresada, documentándose progresivamente hepatoesplenomegalia, transaminitis progresiva, bicitopenia, marcada hiperbilirrubinemia, hiperferritinemia, con incremento de los reactantes de la fase aguda. La biopsia de médula ósea fue concordante con síndrome hemofagocítico. Todos los estudios microbiológicos realizados fueron negativos. No existían datos de enfermedad autoinmune, ni de otras patologías. La paciente fue tratada exclusivamente con dexametasona, con descenso paulatino ulterior de este fármaco. Los estudios de un amplio panel de estudios genéticos fueron negativos.
La paciente fue dada de alta con el diagnóstico de síndrome hemofagocítico de carácter aparentemente idiopático y seguida ulteriormente en consulta sin complicaciones.
CASO 3.-
Varón de 58 años diagnosticado en Sept/2013 por biopsia de médula ósea de LNH T periférico estadio IV-B con cariotipo complejo. Debutó con fiebre, hepatoesplenomegalia, adenopatías paraórticas izquierdas, pancitopenia, infiltración de la médula ósea y del líquido cefalorraquídeo. Los estudios analíticos mostraron hipertrigliceridemia, hiperferritinemia, hipofibrinogenemia, y disminución parcial de expresión de la perforina en las células NK, a los que se unió posteriormente signos de hemofagocitosis en la medula ósea. Todos estos datos sugirieron el diagnóstico de linfohistocitosis hemofagocítica. Por ello, se administró el protocolo LHH-04 combinando dexametasona, etopósido, y ciclosporina, y aplicándose ulteriormente inmunoglobulinas y fibrinógeno por vía intravenosa a causa de inmunodeficiencia e hipofibrinogenemia respectivamente. Una vez obtenido el diagnóstico de linfoma T periférico por biopsia de médula ósea se sustituyó la pauta LHH-04 por quimioterapia combinada tipo ciclofosfamida, adriamicina, vincristina, etopósido, prednisona (CHOEP), aplicándose un total de 5 ciclos y obteniéndose la remisión completa sistémica, pero persistiendo enfermedad en líquido cefaloraquideo. Por ello, como segunda línea se aplicó la combinación metotrexate/arabinosido de citosina a dosis altas. A pesar de los esfuerzos terapéuticos el 28/Feb/2014 se evidenció progresión de la enfermedad con afectación poliadenopática y de otros tejidos. La recidiva fue confirmada histológicamente como linfoma T periférico no especificado. Otras líneas de quimioterapia aplicadas ulteriormente no fueron efectivos y la evolución fue rápida y desfavorable. El diagnóstico fue de síndrome hemofagocítico en relación a linfoma no Hodgkin T periférico.
CASO 4.-
Mujer de 82 años de edad diagnosticada en Mayo/2012 de Leucemia linfática crónica estadio I de Rai. En Oct/2014 fue catalogada de estadio II-de Rai y sometida a una primera línea de tratamiento con Clorambucil y Obinutuzumab. Ante el fracaso terapéutico, se sometió posteriormente a Bendamustina-Rituximab con ausencia de respuesta y como tercera línea a Ibrutinib obteniendo remisión parcial. En esta época mostró un cuadro de anemia hemolítica autoinmune por anticuerpos caliente. A lo largo del 2016 manteniéndose la respuesta al Ibrutinib la enferma desarrolló deterioro del estado general, detectándose por PET-TAC imágenes tumorales en varias localizaciones (pulmón, hígado, tiroides, suprarrenales, hueso y probablemente en médula ósea). El 2/Enero/2017 la paciente ingresa por fiebre, pancitopenia progresiva, cuadro hemolítico, hiponatremia, hallazgos compatibles con síndrome hemofagocítico en la médula ósea y evidencia en PET-TAC de enfermedad multimetastásica. Se hace el diagnóstico de síndrome hemofagocítico y posible tumor asociado. Simultáneamente, se inicia terapia con dexametasona y etopósido consiguiéndose una respuesta parcial y temporal, con empeoramiento ulterior durante la administración de ciclosporina llegando la ferritina 97.000 ng/ml y documentándose posteriormente infección por citomegalovirus, fracaso multiorgánico y estatus epiléptico, con fallecimiento rápido de la paciente.
En síntesis, el diagnóstico fue de leucemia linfática crónica B, anemia hemolítica adquirida autoinmune, posible tumor metastásico no documentado, citomegalovirosis, y síndrome hemofagocítico asociado, con respuesta transitoria al tratamiento y evolución rápidamente fatal.
TRATAMIENTO DE LA LHH EN EL ADULTO
La mayor experiencia terapéutica deviene del ámbito periatrico. En el protocolo auspiciado por la Sociedad Histiocítica conocida como LHH-94 y basado en la combinación de etopósido y dexametasona con la adición posterior de ciclosporina A y metotrexate intratecal, se obtuvo un cambio importante en el pronóstico de la LHH infantil. Una enfermedad inexorablemente fatal en 1-2 meses aproximadamente se convirtió en un síndrome que mostraba frecuentes respuestas al tratamiento citado y que seguido de trasplante alogénico de progenitores hematopoyéticos ofrecía una supervivencia del 54% a los 5 años (14). El trasplante alogénico de progenitores hematopoyéticos se indican en todos los casos de LHH con base genética, historia familiar o enfermedad resistente o recidivante. Un protocolo ulterior, también de la Sociedad Histiocítica, conocida también como LHH-04 caracterizado por la administración inicial de ciclosporina A a la pauta clásica etopósido/dexametasona y aplicación combinada de metotrexate intratecal y prednisona, obtuvo resultados similares, sin apreciarse claras diferencias (15). El enfoque terapéutico y el protocolo LHH-94 se observan en las tablas 3 y 4 respectivamente. Así como la mayoría de los casos LHH primarios o de base genética/familiar, ocurren en la infancia, la mayoría de los casos en adultos son secundarios, no existiendo una base genética aunque sí frecuentemente se asocian a procesos inductores tal como infección, neoplasias, enfermedades reumatológicas, y siendo minoritario el número de pacientes que son catalogados como idiopáticos, es decir no asociados a un proceso definido. No existen guías de tratamiento para el manejo de la LHH del adulto y forzosamente la actitud terapéutica se basa en la experiencia obtenida en pediatría. Por ello, la terapia frecuentemente en los pacientes adultos hasta cierto punto ha de ser personalizada (16,17).


En principio, en la LHH del adulto si se asocia una causa predisponente como infección, neoplasia o trastorno autoinmune, el factor etiológico existente hay que tenerlo en cuenta desde el punto de vista terapéutico. Si como es relativamente común en el adulto la LHH se asocia a linfoma no Hodkgin, frecuentemente T periférico el tratamiento debe de incluir primariamente la terapia anti-linfoma, comúnmente la quimioterapia conocida como CHOP o preferiblemente añadiendo etopósido a esta combinación, régimen conocido como CHOEP. En ausencia de una rápida respuesta en cuestión de días se debe de añadir tratamiento inmunosupresor concomitante con el esquema LHH-94, basado en etopósido/dexametasona. La terapia debe de aplicarse con olvido de las cifras hemoperiféricas y añadiendo terapia de soporte que incluye la administración de trasfusiones, profilaxis antiinfecciosa y factores de crecimiento. La función básica de la adición de etopósido no es solo lograr una inmunosupresión profunda, sino quizá más importante conseguir una marcada linfo-depleción. En caso de disfunción hepática y de fracaso renal es recomendable la disminución de la dosis de este fármaco, con ulterior ascenso si la función hepática y renal mejoran. El etopósido se une a la albúmina. En la LHH es común la marcada hipoalbuminemia con exceso de etopósido libre y por tanto potenciación clínica del mismo. A efectos prácticos no se corrige la dosis con arreglo a la albuminemia. La dexametasona contribuye a la obtención de inmunosupresión y reducción del marcado componente inflamatorio. Dependiendo de la etiología de la LHH del adulto, si no se obtiene respuesta adecuada de manera rápida al tratamiento de primera línea y la causa predisponente es una neoplasia tal como linfoma, leucemia aguda, o mielodisplasia puede estar indicada la práctica de un trasplante alogénico de progenitores hematopoyéticos si se dispone del donante apropiado. El donante ideal es un hermano HLA idéntico. En caso de búsqueda de un donante familiar es imprescindible la práctica de un estudio de mutaciones genéticas antes de aceptar al hermano como donante. Puede utilizarse un hermano en caso de ser portador heterocigoto de una mutación genética relacionada con la función citotóxica. Si no existe la opción de un hermano HLA idéntico, debe de iniciarse la búsqueda de un donante no emparentado HLA idéntico o con una sola diferencia antigénica. En su ausencia un haplotrasplante o el uso de cordón umbilical pueden estar justificados. El momento ideal para la realización del trasplante no es en el seno de enfermedad muy activa con muy evidente hiperinflamación ya que en este supuesto la práctica del trasplante conllevaría más clínica hiperinflamatoria por mayor hipercitoquinemia y mayor tasa de enfermedad injerto contra huésped. En estos pacientes se debería intentar una terapia puente hasta el trasplante, tal como el empleo de alemtuzumab, un antiCD52 que provoca intensa linfodeplección. Los resultados clínicos obtenidos por el trasplante de progenitores hematopoyéticos cuando en lugar del acondicionamiento mieloablativo se han utilizado los regímenes de intensidad reducida, han sido mejores.
Los pacientes catalogados como síndrome de activación macrofágica habitualmente con procesos autoinmunes de base tal como artritis reumatoide juvenil o lupus eritematoso sistémico, se tratan exclusivamente con esteroides, omitiendo el etopósido. En caso de respuesta inadecuada se debe de intensificar el régimen inmunosupresor específico para el proceso de base. Debe de recordarse el carácter mutagénico del etopósido.
Recientemente ha sido aprobado por la FDA un anticuerpo monoclonal IgG 1 que inhibe el interferón gamma tanto para los pacientes pediátricos como para los adultos con LHH resistente, recidivante, progresiva o en los intolerantes a la terapia convencional. La aprobación ha incluido a los pacientes adultos a pesar de la inexistencia de datos clínicos. Por ello parece juicioso que este monoclonal, emapalumab, no sustituya en el adulto a la terapia estándar de primera línea (18).
Los pacientes que no responden al régimen inicial LHH 94, que suponen al menos un 30% del total pueden optar un tratamiento de segunda línea tal como globulina antitimocítica, (inhibidor de la vía JAK) ruxolitinib (19), (anti-rIL1) anakinra y la pauta DEP que incluya la doxorubicina liposomal combinada al etopósido y prednisona.
En síntesis, a pesar de los tratamientos aplicados la mortalidad precoz de la LHH del adulto es alta llegando a cifras cercanas del 50% y aún mayores cerca del 70% cuando se trata de casos asociados de procesos neoplásicos. A la vista de estas cifras, es evidente la necesidad de mejorar la precocidad en el diagnóstico, así como las perspectivas terapéuticas en estos enfermos.
BIBLIOGRAFÍA
- Tong QJ, Godbole MM, Biniwale N, Jamshed S. An elusive diagnosis: case reports of secondary hemophagocytic lymphohistiocytosis and review of current literature. Cureus. 2019; 11(4): e4548.
- Risma KA, Marsh RA. Hemophagocytic lymphohistiocytosis: clinical presentations and diagnosis. J Allergy Clin Inmun. 2019; 7(3): 824-832.
- Schram AM, Berliner N. How I treat hemophagocytic lymphohistiocytosis in the adult patient. Blood. 2015; 125: 2908-2914.
- Machowicz R, Janka G, Wiktor-Jedrejczak W. Similar but not the same: differential diagnosis of HLH and sepsis. Crit Rev Oncol Hemat. 2017; 114: 1-12.
- Jordan MB, Allen CE, Greenberg J, et al. Challenges in the diagnosis of hemophagocytic lymphohistiocytosis: recommendations from the North American Consortium for Histiocytosis (NACHO). Pediatr Blood Cancer. 2019; 66(11): e27929.
- Gurunathan A, Boucher AA, Mark M, et al. Limitations of HLH-2004 criteria in distinguishing malignancy-associated hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2018; 65(12): e27400.
- Trottestam H, Horne A, Aricó M, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. 2011; 118(17): 4577-4584.
- Ehl S, Astigarraga I, von Bahr Greenwood T, et al. Recommendations for the use of etoposide-based therapy and bone marrow transplantation for the treatment of HLH: consensus statements by the HLH Steering Committee of the Histiocyte Society. J Allergy Clin Inmun. 2018; 6(5): 1508-1517.
- Bergsten E, Horne A, Aricó Micó, et al. Confirmed efficacy of etoposide and dexametasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. 2017; 130(25): 2728-2738.
- Marsh RA. Epstein-Barr virus and hemophagocytic Lymphohistiocyto. Front Immunol. 2018. https://doi.org/10.3389/fimmu.2017.01902
- Horne AC, Wickström R, Jordan MB, et al. How to treat involvement of the central nervous system in hemophagocytic lymphohistiocytosis? Curr Treat Options Neurol. 2017; 19: e3.
- Hayden A, Park S, Giustini D, Lee AYY, Chen LYC. Hemophagocytic syndromes (HPSs) including hemophagocytic lymphohistiocytosis (HLH) in adults: a systematic scoping review. Blood Rev. 2016; 30(6): 411-420.
- Hayden A, Lin M, Park S, et al. Soluble interleukin-2 receptor is a sensitive diagnostic test in adult HLH. Blood adv. 2017; 1(26): 2529-2534.
- Marsh RA, Haddad E. How I treat primary haemophagocytic lymphohistiocytosis. Br J Haematol. 2018; 182(2): 185-199.
- Wang Y, Wang Z. Treatment of hemophagocytic lymphohistiocytosis. Curr Opin Hematol. 2017; 24(1): 54-58.
- Daver N, McClain K, Allen CE, et al. A consensus review on malignancy-associated hemophagocytic lymphohistiocytosis in adults. Cancer. 2017; 123: 3229-3240.
- La Rosée P, Horne AC, Hines M, et al. Recommendations for the management of hemophagocytic lymphohistocytosis in adults. Blood. 2019; 133(23): 2465-2477.
- Vallurupalli M, Berliner N. Emapalumab for the treatment of relapsed/refractory hemophagocytic lymphohistiocytosis. Blood. 2019; 134(21): 1783-1786.
- Ahmed A, Merrill SA, Alsawah F, et al. Ruxolitinib in adult patients with secondary haemophagocytic lymphohistiocytosis: an open-label, single-centre, pilot trial. Lancet Haematol. 2019; 6(12): e630-e637.
DECLARACIÓN DE TRANSPARENCIA
El autor/a de este artículo declara no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
ranm tv
José María Fernández-Rañada y de la Gándara
Real Academia Nacional de Medicina de España
C/ Arrieta, 12 · 28013 Madrid
Tlf.: +34 91 547 03 18 | E-Mail: jmranada@yahoo.es
Año 2021 · número 138 (01) · páginas 24 a 30
Enviado*: 16.02.21
Revisado: 25.02.21
Aceptado: 15.03.21
* Fecha de lectura en la RANM