

Resumen
La Hipertensión Arterial pulmonar es un síndrome central producido por un gran número de enfermedades cardiológicas, pulmonares, y enfermedades sistémicas que repercuten dañando el lecho pulmonar. Se define por la existencia de una presión sistólica de arteria pulmonar superior a 30 o presión media superior a 25 mmHg. Este criterio de definición se mantiene desde hace más de 60 años. Sin embargo la clasificación actual recoge dos conceptos: una Hipertensión Arterial Pulmonar (HAP) con dos grupos de trastornos en las que se elevan solamente las resistencias arteriales pulmonares y cinco grupos que se clasifican como Hipertensión Pulmonar (HP): HP Secundaria a Enfermedad venooclusiva pulmonar, HP secundarias a enfermedades del lado izquierdo del corazón; HP Enfermedades obliterativas e hipoxemia pulmonar.; HP Enfermedades trombo oclusivas pulmonares, y un grupo de HP multifactorial. La diferencia se encuentra en cómo las distintas enfermedades afectan el lecho vascular pulmonar, y como alteran la fisiología o comportamiento de las resistencias pulmonares que son los conceptos que se deben manejar al hablar de este síndrome y cuyos cambios estructurales y manejo expondremos en un artículo posterior.
Abstract
Pulmonary Arterial Hypertension is a central syndrome produced by a large number of cardiological, pulmonary, and systemic diseases that affect the lung bed. It is defined by the existence of a pulmonary artery systolic pressure greater than 30 or a mean pressure greater than 25 mmHg. This definition criterion has been maintained for more than 60 years. However, the current classification includes two concepts: a Pulmonary Arterial Hypertension (PAH) with two groups of disorders in which only pulmonary arterial resistance increases and five groups that are classified as Pulmonary Hypertension (PH): PH Secondary to Pulmonary Veno-occlusive Disease , HP secondary to diseases of the left side of the heart; HP Obliterative diseases and pulmonary hypoxemia; HP Pulmonary thrombus occlusive diseases, and a group of multifactorial HP. The difference is found in how the different diseases affect the pulmonary vascular bed, and how they alter the physiology or behavior of pulmonary resistance, which are the concepts that must be handled when talking about this syndrome and whose structural changes and management we will discuss in a later article.
Palabras clave: Hipertensión Arterial Pulmonar; Hipertensión Pulmonar; Resistencias pulmonares totales; Resistencias pulmonares venosas; Resistencias pulmonares arteriales.
Keywords: Pulmonary Arterial Hypertension; Pulmonary hypertension; Total Pulmonary Resistances; Venous pulmonary resistances; Pulmonary arterial resistance.
ÍNDICE DE ABREVIATURAS
5-HT: 5 hidroxi triptamina.
AI: Aurícula izquierda
AMPc: Adenosín monofosfato cíclico.
AP: Arteria Pulmonar
Ca++: Calcio
CLM: Célula lisa Muscular
CO: Monóxido de carbono
Cp: Capilar pulmonar
EPOC: Enfermedad pulmonar obstructiva crónica
ET-1: endotelina-1
EVOP: Enfermedad vascular oclusiva pulmonar.
GMPc: Guanosín monofosfato cíclico
HAP: Hipertensión Arterial Pulmonar
HP: Hipertensión Pulmonar.
HPTC: HP iromboembólica crónica
NO: óxido nítrico
PAP: Presión Arteria Pulmonar.
PAPm: Presión Arteria Pulmonar media
PCP: Presión Capilar Pulmonar
PD: Presión diastólica
PGI2: Prostaciclina
Ps: Presión sistólica.
PTDVI: Presión telediastólica del Ventrículo izquierdo
R: Resistencias
RAP: Resistencias arteriales pulmonares.
RPA: Resistencias pulmonares arteriales
RPT: Resistencias Pulmonares Totales
RPV: Resistencias pulmonares venosas
RVP: Resistencias Venosas pulmonares
TxA2: tromboxano A2
VPs: venas pulmonares
V: Volumen
1. INTRODUCCIÓN
El lecho vascular pulmonar presenta bajas resistencias vasculares, gran distensibilidad y una gran reserva funcional, capaz de aumentar varias veces el flujo durante el ejercicio (1). Un gran número de enfermedades cardiológicas, pulmonares y algunas enfermedades sistémicas afectan al pulmón produciendo cambios en los vasos pulmonares con vasoconstricción y trastornos proliferativos que producen aumento de las resistencias que obligan a que se eleve la Presión de Arteria Pulmonar (AP) por encima del nivel normal de 30 mmHg de presión sistólica (Ps) ó 25 mmHg de Presión media (Pm). Estos límites definen la Hipertensión Pulmonar (HP). Cada vez se conocen mejor las molécuias y mecanismos que mantienen el tono vascular, constrictor / dilatador y responsables de iniciar la proliferación celular, responsable de los cambios anatomopatológicos en la estructura vascular, que describiremos en una segunda parte de este trabajo docente. Este conocimiento ha permitido un mejor diagnóstico y un notable avance en el tratamiento, que ha mejorado el pobre pronóstico que presentan los pacientes con HP.
2. Un recuerdo de cómo se comportan las presiones pulmonares
La presión pulmonar está condicionada por el flujo o Volumen (V) y las resistencias pulmonares totales (RPT) (P=VxR).
El flujo pulmonar y el sistémico son iguales en condiciones normales (sin cortocircuitos). Las RPT son la suma de las resistencias pulmonares venosas (RVP ó RPV) y las arteriales (RAP ó RPA). La presión diastólica final o telediastólica del Ventrículo izquierdo (PTDVI) en condiciones normales no supera los 15 mmHg. Esta presión se transmite a la aurícula izquierda (AI), venas pulmonares (VPs), lecho capilar pulmonar (CP) y a la presión diastólica (Pd) de Arteria Pulmonar (AP), siendo igual en todos estos tramos vasculares.
La presión sistólica normal de la AP no supera 30 mmHg. La presión media (PmAP) se calcula como la PS sumada a dos veces la presión diastólica (PD), dividido por tres (30 + 15×2/ 3 = 20). (Figura 1)
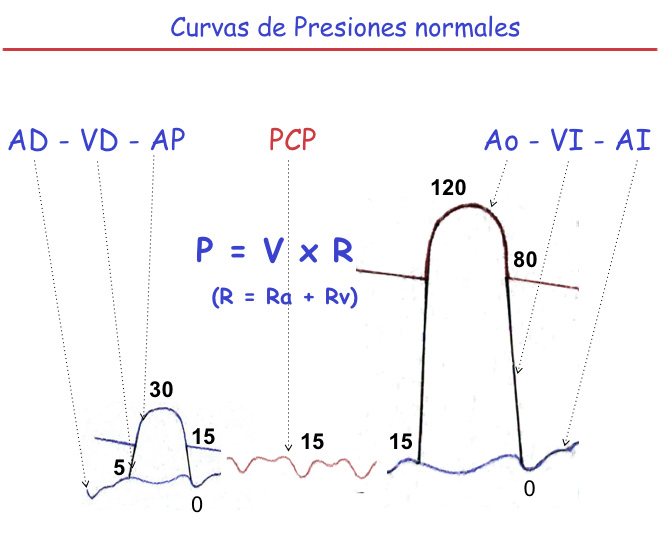
La RPT, calculada en unidades simples, es la PAP en mmHg dividida por el flujo pulmonar en litros por minuto. Si a la PAPm le restamos la Presión Capilar media (PCP) o presión venosa, el cálculo nos da las resistencias de la parte arterial o precapilar (RAP) de la circulación pulmonar. Restando éstas a las resistencias totales calculamos las RVP o postcapilares. La Pm de AP (20 mm Hg) es ligeramente superior a la PCP (15 mmHg). Esa diferencia entre presión media de AP y la PCP expresa las RAP, muy pequeñas en la circulación pulmonar normal, cuando la Pd de AP es igual a la PCP. Pero conforme más se amplía la diferencia entre la PdAP y la PCP, más elevadas son las RAP, más elevado es el gradiente entre la AP y el CP producido por la reducción de la luz de las arteriolas pulmonares.
En la HP por una enfermedad en las cavidades izquierdas o en las venas pulmonares se elevarán las RVP que contribuyen al aumento de las RPT. Paralelamente la Pd de la AP tendrá la misma presión elevada que el capilar pulmonar. Si la PCP está elevada, y la Pd de la AP es aún más alta que la PCP, la RPV estarán elevadas (PCP) , pero también lo estarán las RPA, y por tanto las RPT.
En el resto de las patologías que producen HAP, se elevan solamente las RAP y permanecen normales las RPV (la PCP).
3. Hipertensión Pulmonar (HP)
La Hipertensión Pulmonar (HP) se define por un aumento de la la Presión de Arteria Pulmonar (PAP) por encima del nivel normal de 30 mmHg de presión sistólica (Ps) ó 25 mmHg de Presión media (Pm). Desde el punto de vista hemodinámico la HP puede clasificarse en HP ligera entre 30 y 50 mmHg de Ps Ap y de 25 a 40 mmHg de PAP media. Moderada entre de 50 – 70 mmHg de Ps, con PAPm de 40 a 55 mmHg, e HP severa con Ps superior a 70 mmHg, y a 55 mmHg de PAPm.
4. Clasificación de la Hipertensión arterial pulmonar
4.1. En 1958 Paul Wood definió la HAP por el aumento de la presión pulmonar por encima del límite de 30/15 que hemos explicado y realizó una clasificación fisiopatológica de este trastorno. Describió siete categorías de HAP: Pasiva, Hipercinética, Obstructiva, Obliterativa, Vasoconstrictiva, Poligénica (o mixta) y Reactiva. (2)
4.1- 1.- HAP pasiva o venosa: producida por aumento de las RPV o lo que es lo mismo, la PCP. Se elevan en todas las cardiopatías izquierdas, incluida la trombosis venosa pulmonar, la pericarditis constrictiva y el derrame pericárdico a tensión o taponamiento cardiaco.
4.1-2.- HAP hipercinética: por aumento del flujo pulmonar, superior al sistémico, por cortocircuito izquierda derecha, que acompaña a cardiopatías congénitas cianóticas o no cianóticas y a fístulas arteriovenosas con alto flujo. La PCP es normal. Los cortocircuitos a nivel ventricular (comunicación interventricular) o de grandes arterias (persistencia del conducto arterioso) con un flujo pulmonar superior a dos veces el sistémico (grandes), son cortocircuitos de alta presión y alto flujo, y desarrollan precozmente HAP por aumento de las RPA. Las comunicaciones interauriculares (CIA) tardan en desarrollar cortocircuitos con mucho flujo y baja presión y desarrollan la HAP por aumento de RPA en la 5ª o 6ª década de la vida. Esta evolución de esta HP recibe también el nombre de síndrome de Eissenmenger.
4.1-3.- HAP obstructiva: resultado de una embolia masiva, embolismos múltiples o trombosis pulmonar, puede ser aguda, subaguda o crónica. También la puede originar la ligadura quirúrgica de una AP principal, en casos de neumectomía, ausencia congénita, estenosis o compresión de ramas pulmonares. Se elevan solo las RPA.
4.1-4.- HAP obliterativa: ocurre en enfermedades pulmonares como el enfisema y la fibrosis intersticial. También en las vasculitis o endarteritis de grandes o pequeños vasos pulmonares. Se elevan las RPA. Las RPV son normales. En la clasificación de Wood se incluía en esta categoría la HAP primaria.
4.1-5.- HAP vasoconstrictiva en respuesta a la hipoxia alveolar aguda o crónica, complicando infecciones respiratorias agudas y enfermedades pulmonares crónicas. Se elevan las RPA.
4.1-6.- HAP poligénica, aparece en una de las formas anteriores: pasiva, hipercinética u obliterativa y se complican con los factores vasoconstrictores hipóxicos que acompañan a un enfisema pulmonar o una trombosis pulmonar masiva. Se elevan las RPA.
4.1-7.- HAP reactiva acompaña a la HAP venosa o pasiva, y a la hipercinética. En las primeras fases se inicia con una vasoconstricción reversible. Paulatinamente los cambios anatomopatológicos llegan a ser irreversibles y conducen al fallo derecho terminal.
A partir de esta clasificación y del mejor conocimiento de las enfermedades que producen el aumento de la PAP se han realizado sucesivas clasificaciones. (3)
4.2 Las nuevas clasificaciones diferencian Hipertensión Pulmonar (HP) de Hipertensión Arterial pulmonar (HAP). Siendo críticos, la hipertensión pulmonar es siempre arterial, por lo que una separación terminológica como la que proponen estas nuevas clasificaciones no deja de ser artificiosa. En la tabla 1, tomada de Galie et al. (4) aparece la clasificación del año 2015 con ambas entidades HAP e HP.
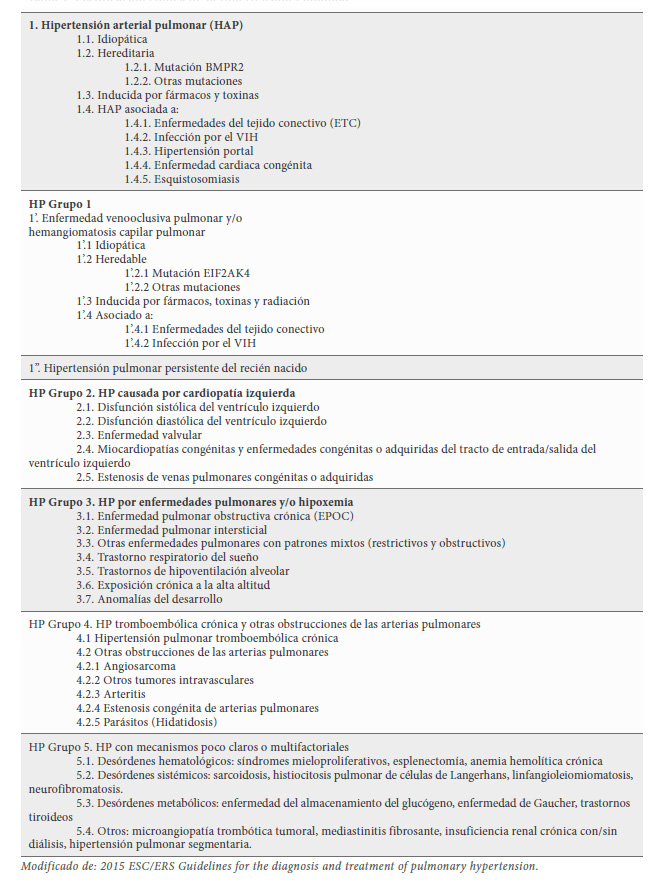
La HP y la HAP tienen el mismo criterio diagnóstico, el nivel de presión, PsAP superior a 30 mmHg y PAPm es superior a 25 mmHg y la distinción está en el comportamiento de las Resistencias Pulmonares (RP) y la causa que la produce. El diagnóstico estrictamente depende de que se realice un cateterismo cardiaco derecho, pero en la práctica se mide y diagnostica durante la reaiización de un ECOCG según el valor obtenido del gradiente Ventrículo Derecho (VD) a Aurícula Derecha (AD). La alternativa a la misma podría hacerse emplenado indistintamente cualquiera de los dos términos, definiendo la enfermedad causal y cuáles son las Resistencias Pulmonares que están elevadas, las totales (siempre), las venosas, (en los fallos izquierdos) y las arteriolares en el resto, salvo en las hipercinéticas en que estará elevada la presión y el flujo antes de producirse la HP reactiva.
Siguiendo las nuevas clasificaciones, la primera categoría, la Hipertensión Arterial Pulmonar (HAP), se caracteriza porque la hipertensión es precapilar, con RAP superiores a 3 unidades Wood, siendo la presión venosa, PCP o de enclavamiento (RVP), normal, inferior a 15 mmHg. La HAP incluye cuatro subgrupos: Idiopática, Hereditaria, por fármacos, y asociadas a otras enfermedades. Comparten un cuadro clínico similar y cambios patológicos prácticamente idénticos sobre la microcirculación pulmonar. Las diferencias están en la patología original. (5)
Las patologías bajo el epígrafe de HP se agrupan en cinco grupos:
4.2-1 Grupo 1 que está integrada por la Enfermedad venooclusiva pulmonar, que puede tener un origen idiopático, hereditario, inducido por fármacos o asociada a enfermedades del tejido conectivo o al VIH. Un quinto subgrupo: (1”) corresponde a la HP persistente del recién nacido. En estas enfermedades están elevadas principalmente las RPV, como parte fundamental en la elevación de las RPT.
4.2-2 Grupo 2, HP por cardiopatía izquierda (o HAP pasiva o venosa de la clasificación de Wood). En su inicio las RPT están elevadas a expensas de las RPV. Dejadas a su evolución producirán cambios reactivos que producirá elevación también de las RPA.
4.2-3 Grupo 3. HP por enfermedades pulmonares y/o hipoxemia. (o HAP obliterativa en la clasificación de Wood). La presión se eleva por aumento de las RPA, pero el dato distintivo está en la enfermedad que lo origina.
4.2-4 Grupo 4: HP tromboembólica crónica (HPTC) y otras obstrucciones de las arterias pulmonares. (o HP obstructiva en la clasificación de Wood). Ocurre como en el grupo anterior por elevación de las RPA. No hay elevación de RPV.
4.2-5 Grupo 5. HP con mecanismos poco claros o multifactoriales en este grupo se incluyen trastornos hematológicos, enfermedades sistémicas, como la sarcoidosis, trastornos metabólicos por enfermedad de depósito como el Gaucher y otras enfermedades, como la microangiopatía trombótica tumoral y otras enfermedades de etiología multifactorial. Desde el punto de vista de la afectación pulmonar, es principalmente precapilar o por aumento de RPA.
5. Epidemiología
Es complicado fijar la cifra de hipertensos pulmonares, dado que la causa más frecuente y común de este trastorno son las cardiopatías del lado izquierdo, seguidas de la que acompaña a las enfermedades parenquimatosas pulmonares. Pero en el grupo 1 de HAP (precapilar que no corresponde a enfermedades parenquimatosas pulmonares) incluida la forma de HAP idiopática se han estimado cifras muy variables entre 2,4 y hasta 60 casos por millón de habitantes(6,7)
En el Grupo 2 de HP puede presentarse en más del 60% de pacientes con Insuficiencia cardiaca congestiva o valvulopatías izquierdas en fases avanzadas de la enfermedad sin reparación adecuada.
En el grupo Grupo 3 de HP en pacientes con enfermedad pulmonar obstructiva crónica (EPOC) y con patología intersticial, desarrollan HP frecuente de grado leve a moderado, mientras que el grupo pacientes con síndrome combinado de enfisema más fibrosis pulmonar, desarrollan HP severa de forma habitual y condiciona un peor pronóstico de la enfermedad.
En el Grupo 4, los que presentan HPTC la prevalencia que se señala es de 3,2 casos/1.000.000 habitantes, y una incidencia de 0,9/1.000.000/año. En pacientes con trastornos trombofílicos, como pueden ser el anticoagulante lúpico y anticuerpos antifosfolipídicos, existe HP hasta en un 31,9% de estos pacientes.
En un estudio de un laboratorio de ecocardiografía la prevalencia de la HP en todos los pacientes fue del 10,5%. El 78,7% padecían cardiopatía izquierda (grupo 2), el 9,7% sufría enfermedades pulmonares e hipoxemia (grupo 3), el 0,6% tuvo HPTC (grupo 4) y un 4,2% tenía HAP. No fue posible definir el diagnóstico del 6,8% restante. (8)
6. genética
Se han descrito algunas mutaciones genéticas directamente relacionadas con HAP. La mutación más conocida es la del gen BMPR2 descrita hasta en el 75% de los casos de HAP familiar y en el 25% de los casos con HAP esporádica (Grupo 1 HP). Este gen BMPR2 codifica un receptor implicado en la proliferación celular vascular. En pacientes con enfermedad veno oclusiva pulmonar (EVOP) y con hemangiomatosis capilar pulmonar, aparece una mutación en el gen EIF2AK4 (9).
Estos tipos de HP, presentan cambios estructurales comunes en todos ellos, más los característicos de cada patología causal que tienen criterios diagnósticos y pruebas complementarias que permiten un diagnóstico sindrómico y causal. Con ello se puede estructurar un tratamiento cada vez más eficaz, que se expondrá en el siguiente artículo docente sobre Hipertensión pulmonar que se publicará en Anales de la RANME.
BIBLIOGRAFÍA
- Domingo E, Grignola JC, Aguilar R et al. Impairment of pulmonary vascular reserve and right ventricular systolic reserve in pulmonary arterial hypertension. BMC Pulmonary Medicine 2014, 14:69. http://www.biomedcentral.com/1471-2466/14/69
- Wood P. Pulmonary hypertension with special reference to the vasoconstrictive factor. Br Heart J. 1958; 20: 557-570)
- Martín-Suñé N, Ríos-Blanco JJ. Afectación pulmonar de las vasculitis. Arch Bronconeumol. 2012; 48: 410-418
- Simonneau G, Galiè N, Rubin LJ, Langleben D, Seeger W, Domenighetti G, Gibbs S, Lebrec D, Speich R, Beghetti M. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004;43(Suppl 1):S5–S12.)
- Galié N, Humbert M, Vachiery JL, Gibbs JS, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, et al Guía ESC/ERS 2015 sobre diagnóstico y tratamiento de la hipertensión pulmonar. Rev Esp Cardiol. 2016; 69(2): 177.e1-e62
- Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2019; 53: 1801913 [https://doi.org/10.1183/13993003.01913-2018].
- Peacock AJ, Murphy NF, McMurray JJV, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J 2007;30:104–109.
- McGoon MD, Benza RL, Escribano-Subias P, Jiang X, Miller DP, Peacock AJ, Pepke-Zaba J, Pulido T, Rich S, Rosenkranz S, Suissa S, Humbert M. Pulmonary arterial hypertension: epidemiology and registries. J Am Coll Cardiol 2013; 62(Suppl):D51–D59.
- Gabbay E, Yeow W, Playford D. Pulmonary arterial hypertension (PAH) is an uncommon cause of pulmonary hypertension (PH) in an unselected population: the Armadale echocardiography study. Am J Resp Crit Care Med. 2007;175:A713.
- Best DH, Austin ED, Chung WK, Elliott CG. Genetics of pulmonary hypertension. Curr Opin Cardiol. 2014;29:520-7. doi: 10.1097/ HCO. 0000000000000105. PMID: 25159282.
DECLARACIÓN DE TRANSPARENCIA
El autor/a de este artículo declara no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
José Ramón de Berrazueta Fernández
Real Academia Nacional de Medicina de España
C/ Arrieta, 12 · 28013 Madrid
Tlf.:+34 91 547 03 18 | E-Mail: joseramon@berrazueta.com
An RANM. 2021;138(02): 137 – 142
Enviado: 01.08.21
Revisado: 07.08.21
Aceptado: 26.08.21