

Resumen
La epilepsia es una enfermedad neurológica crónica caracterizada por la aparición espontánea y recurrente de crisis epilépticas, consecuencia de una actividad neuronal anómala, excesiva y sincrónica en el cerebro. La epilepsia del lóbulo temporal (ELT) es la forma más común de epilepsia focal en adultos estando caracterizada por esclerosis hipocampal, gliosis reactiva, neurodegeneración y reorganización sináptica. Los modelos animales de ELT basados en la administración de agentes convulsivantes desencadenan un status epilépticus (SE) que progresa hacia la aparición de crisis epilépticas recurrentes espontáneas. Entre éstos, se encuentran los inducidos por la administración sistémica de pilocarpina o por inyección intrahipocampal de ácido kaínico que se caracterizan por 3 fases claramente definidas: (i) crisis aguda de SE; (ii) periodo latente y (iii) aparición de crisis espontáneas recurrentes. Estos modelos no sólo reproducen la mayoría de las características neuropatológicas de la ELT sino que permiten identificar biomarcadores de epileptogénesis y potenciales dianas farmacológicas. El uso de técnicas de neuroimagen como la tomografía por emisión de positrones (PET) con el radiotrazador 18F-Fluorodesoxiglucosa (18F-FDG) identifica hipometabolismo cerebral en el periodo latente que no sólo localiza el foco epiléptico sino que es un biomarcador de diagnóstico precoz. Otras técnicas de neuroimagen permiten detectar entre otros, biomarcadores de neuroinflamación, de alteraciones en la permeabilidad de la barrera hematoencefálica y de activación astrocitaria, todos ellos asociados a la epileptogénesis. Por último, el uso de la quimiogenética mediante tecnología DREADDs (Designer Receptors Exclusively Activated by Designer Drugs) en modelos murinos permite modular la actividad astrocitaria siendo una herramienta novedosa que considera la contribución del papel de los astrocitos en las alteraciones metabólicas cerebrales en la epileptogénesis.
Abstract
Epilepsy is a chronic neurological disease characterized by spontaneous recurrently occurring epileptic seizures as a consequence of abnormal, excessive and synchronous neuronal activity in the brain. Temporal lobe epilepsy (TLE) is the most common form of focal epilepsy in adults, being characterized by hippocampal sclerosis, reactive gliosis, neurodegeneration, and synaptic reorganization. Animal models of TLE based on the administration of convulsive agents trigger a status epilepticus (SE) that progresses towards the occurrence of spontaneous recurrent seizures. Among these models are those induced by the systemic administration of pilocarpine or by intrahippocampal injection of kainic acid, both being characterized by 3 clearly defined phases: (i) acute SE seizures; (ii) latent period and (iii) occurrence of recurrent spontaneous seizures. These models not only reproduce most of the neuropathological TLE features but also allow for the identification of biomarkers of epileptogenesis and potential pharmacological targets. The use of neuroimaging techniques such as positron emission tomography (PET) with the radiotracer 18F-Fluorodeoxyglucose (18F-FDG) identifies brain hypometabolism in the latent period that not only localizes the epileptic focus but is also an biomarker of early diagnosis. Other neuroimaging techniques allow for detecting, among others, biomarkers of neuroinflammation, alterations in the permeability of the blood-brain barrier and astrocytic activation, all of them associated with epileptogenesis. Finally, the use of chemogenetics through DREADDs (Designer Receptors Exclusively Activated by Designer Drugs) technology in murine models leads to targeted modulation of astrocytic activity, being a novel tool that considers the contribution of the astrocytes role in brain metabolic alterations in epileptogenesis.
Palabras clave: Epileptogénesis; Hipometabolismo; Tomografía por emisión de positrones; Activación glial; DREADDs.
Keywords: Epileptogenesis; Hypometabolism; Positron emission tomography; Glia reactivity; DREADDs.
1. EPILEPSIA Y EPILEPTOGÉNESIS
La epilepsia es una enfermedad neurológica crónica y heterogénea caracterizada clínicamente por una predisposición a padecer crisis epilépticas recurrentes, entendiendo éstas como episodios paroxísticos de signos y/o síntomas provocados por una actividad neuronal anómala, excesiva y sincrónica en el cerebro (1). Tiene una prevalencia aproximada a nivel mundial del 0.5 al 2%. Cada año se diagnostican unos 2,4 millones de nuevos casos. En Europa hay 6 millones de pacientes diagnosticados con epilepsia suponiendo un coste económico estimado de 15.500 millones € anuales para el sistema sanitario. En España se estima que hay 400.000 pacientes, siendo una de las enfermedades que más afecta a la calidad de vida del paciente según la Sociedad Española de Neurología (https://www.sen.es/saladeprensa/pdf/Link270.pdf). Este manuscrito tiene como objetivo revisar el proceso de epileptogénesis mediante el uso de técnicas de neuroimagen en modelos animales. Además, se presentan resultados de nuestro laboratorio que sugieren un papel homeostático de la glía en las alteraciones metabólicas que acompañan a la epileptogénesis.
Las crisis epilépticas se clasifican en base a diferentes características. Así, según su inicio, se clasifican en crisis focales, generalizadas o de inicio no determinado. Dependiendo del área cerebral afectada y la sintomatología predominante, las crisis pueden ser motoras, sensoriales, autonómicas o psíquicas. Atendiendo a su etiología, las crisis pueden ser genéticas, secundarias a una lesión estructural, infecciosas, metabólicas, inmunes o idiopáticas si no existe una causa definida. La distribución por edades de la incidencia de las crisis epilépticas presenta un máximo en la primera infancia y otro en la vejez (2). En el periodo neonatal y primera infancia, las causas potenciales incluyen los traumatismos, infecciones, encefalopatías hipóxico-isquémicas, anomalías congénitas, trastornos metabólicos y convulsiones febriles. La epilepsia del lóbulo temporal (ELT), junto con otros síndromes epilépticos idiopáticos o genéticos, suele comenzar en la infancia y en la adolescencia. Los traumatismos cerebrales son una causa frecuente de la ELT en el adulto. En la vejez, además de los traumatismos cerebrales, las enfermedades cerebrovasculares, los tumores y las enfermedades neurodegenerativas son las causas más frecuentes de la aparición de crisis epilépticas.
Algunas formas de epilepsia se manifiestan meses o años después de un insulto inicial (por ejemplo, tras un traumatismo cerebral o un ictus), o como consecuencia de un defecto genético como la esclerosis tuberosa. Durante el periodo de latencia que transcurre entre el daño inicial y la aparición de crisis epilépticas espontáneas, al menos para las epilepsias adquiridas, se producen cambios metabólicos, neuroinflamación, aumento de la permeabilidad de la barrera hematoencefálica (BHE), cambios sinápticos y neurodegenerativos. Este proceso por el cual los cambios estructurales y funcionales transforman un cerebro normal en uno capaz de generar crisis epilépticas de forma espontánea se conoce como epileptogénesis e incluye la progresión de la enfermedad inducida por el daño desencadenante epileptogénico. El proceso de epileptogénesis implica tres fases: (i) un insulto inicial, lesión o evento precipitante; (ii) un período “latente” (o silente) durante el cual los cambios ocurren como consecuencia de la lesión precedente, transformando así el cerebro normal en un cerebro epiléptico y (iii) después de que la epilepsia ha comenzado, nuevos cambios causan o favorecen la progresión de la enfermedad y de su severidad (3). En este contexto, los estudios longitudinales que implementan las técnicas de neuroimagen permiten identificar y evaluar el área del cerebro afectada, lo que da acceso a intervenir, durante el periodo de latencia, previniendo y/o reduciendo de esta forma los riesgos intrínsecos al proceso de epileptogénesis (4). Teniendo en consideración que alrededor de 27 millones de traumatismos cerebrales se producen al año en el mundo y que el 15% de éstos resultan en epileptogénesis, la identificación y tratamiento eficaz de estos pacientes supondría que 4 millones de casos de epilepsia se podrían evitar al año. Sin embargo, todavía no se dispone de biomarcadores específicos para la epileptogénesis y aunque los tratamientos con fármacos antiepilépticos son capaces de controlar las crisis en la mayoría de los pacientes, alrededor del 30-40% de éstos son refractarios a los tratamientos farmacológicos (5). A pesar de los avances significativos en el diagnóstico y tratamiento de la enfermedad, los datos epidemiológicos no muestran una reducción ni en la aparición de nuevos casos ni en el porcentaje de pacientes que son refractarios a los tratamientos farmacológicos (6).
2. EPILEPSIA DEL LÓBULO TEMPORAL (ELT)
A pesar de que la epilepsia es un síndrome y por lo tanto está lejos de ser una enfermedad única y bien caracterizada, la ELT es la forma más común de epilepsia localizada en adultos. Una de las principales características de ELT es la esclerosis del hipocampo, caracterizada por gliosis reactiva, pérdida neuronal y reorganización sináptica. En las áreas esclerotizadas, la glía (incluyendo microglía y astroglía) experimenta cambios fisiológicos y moleculares que se consideran contribuyen al desarrollo de una red crónicamente hiperexcitable que sustenta las crisis epilépticas recurrentes espontáneas asociadas a la epilepsia.
El diagnóstico de la ELT se alcanza a través de la semiología de las crisis y su confirmación con la electroencefalografía (EEG) y las técnicas de neuroimagen. La descarga epileptiforme en el registro EEG intercrítico es una prueba diagnóstica de alta fiabilidad y la presencia de descargas ictales confirman el diagnóstico. Las técnicas diagnósticas de imagen por tomografía por emisión de positrones (PET, acrónimo de inglés Positron Emission Tomography) localiza la zona epileptógena como un área hipometabólica en la fase interictal, y la imagen por resonancia magnética (MRI, acrónimo inglés de Magnetic Resonance Imaging) localizan la esclerosis del hipocampo. Una mayoría de pacientes con ELT son refractarios al tratamiento farmacológico y por ello las alternativas terapéuticas son la cirugía, la estimulación del vago u otras estimulaciones cerebrales profundas.
3. MODELOS EXPERIMENTALES DE EPILEPSIA DEL LÓBULO TEMPORAL (ELT) Y DE CONVULSIONES AGUDAS
Una de las dificultades tanto práctica como ética en el desarrollo de terapias antiepilépticas es la selección de los pacientes participantes en los ensayos clínicos dado que, sólo un pequeño porcentaje de los sujetos que padecen una lesión cerebral traumática o un insulto potencialmente epileptógeno terminará desarrollando epilepsia. Lamentablemente, en la actualidad no se dispone de biomarcadores de epileptogénesis, que permitieran identificar a aquellos individuos con mayor riesgo de desarrollar epilepsia tras una lesión cerebral o que fueran sensibles a la respuesta de los tratamientos antiepilépticos (7). Es por ello por lo que los programas de investigación en epilepsia tienen como principal reto y objetivo identificar biomarcadores de epileptogénesis. En este contexto, el uso de modelos animales es una herramienta útil para explorar el proceso de epileptogénesis e identificar biomarcadores que puedan ser fácilmente trasladados a la clínica con un objetivo doble: (i) la selección de individuos con una alta probabilidad de desarrollar epilepsia y (ii) la capacidad de evaluar la efectividad de potenciales tratamientos que frenen este proceso.
3.1. Modelos de crisis epilépticas recurrentes espontáneas
Los modelos animales de ELT se basan en la administración de agentes convulsivantes capaces de desencadenar un status epilepticus (SE) y que posteriormente progresará hasta la aparición de crisis epilépticas recurrentes espontáneas, reproduciendo en el animal experimental la mayoría de las principales características neuropatológicas que ocurren en los pacientes con ELT (8-10). A través de los modelos experimentales podemos analizar el proceso de epileptogénesis, inicio y progresión, evaluar los mecanismos moleculares y fisiopatológicos implicados, identificar biomarcadores, dianas terapéuticas, y estudiar los aspectos farmacodinámicos, farmacocinéticos, toxicológicos, con el objetivo final de establecer la eficacia y seguridad de potenciales tratamientos.
El modelo de epilepsia inducido por kindling eléctrico (estimulación eléctrica local en la amígdala o en el hipocampo) inicialmente genera crisis focales que posteriormente se generalizan resultando en la aparición de crisis convulsivas recurrentes. Este modelo, junto con los modelos experimentales de epilepsia inducidos por la administración de ácido kaínico (AK) o de pilocarpina, no sólo producen un SE que progresa a crisis convulsivas recurrentes, sino que son modelos animales fenotípicamente similares a la ELT con esclerosis del hipocampo.
El modelo de epilepsia inducido por AK se basa en su acción sobre los receptores glutamatérgicos ionotrópicos de kainato que modulan la liberación de glutamato en el hipocampo. La administración del AK genera crisis focales secundariamente generalizadas que conducen a la muerte celular por excitotoxicidad, particularmente en la región CA3 del hipocampo (11). De hecho, la inyección de AK es un modelo animal de ELT y de SE bien caracterizado y ampliamente utilizado (12).
La pilocarpina es un alcaloide con acción colinérgica, actuando principalmente a nivel de los receptores muscarínicos M1 y posee efectos convulsivos. El efecto colinérgico se potencia con la administración previa de litio permitiendo así el uso de dosis más bajas de pilocarpina lo que reduce la mortalidad asociada al modelo experimental. Sus efectos son dependientes de la dosis y de la edad del animal. En las ratas jóvenes, la administración de litio-pilocarpina genera un SE que después de un periodo de latencia (semanas o meses) evoluciona en la aparición de crisis espontáneas.
3.2. Modelos de convulsiones agudas
La inyección intra-hipocampal de 4-aminopiridina (4-AP), agente que bloquea los canales de K+, desencadena una despolarización generalizada, causa crisis convulsivas agudas con consecuencias neurológicas permanentes. Otro agente convulsivo es el pentilentetrazol (PTZ), un antagonista no competitivo de los receptores GABAA que administrado a dosis altas genera crisis generalizadas en función de la edad del animal. A diferencia de los modelos de ELT, estos no progresan a crisis espontáneas recurrentes. No obstante, ambos compuestos se emplean en la generación de modelos animales de convulsiones agudas para la evaluación de potenciales fármacos antiepilépticos.
4. BIOMARCADORES DE LA EPILEPTOGÉNESIS
Los biomarcadores ideales, además de ser específicos y fiables, deben ser no invasivos y fácilmente trasladables a la clínica. Más aún, probablemente se requiera de la combinación de diferentes biomarcadores lo que implica un análisis de un elevado número de datos, técnicas avanzadas de estadística e inteligencia artificial en la extracción de patrones de los biomarcadores en su conjunto.
En la tabla 1 se relacionan algunos biomarcadores potencialmente trasladables a la práctica clínica. Los biomarcadores moleculares de fluidos biológicos tienen una gran ventaja debido a su accesibilidad y su cuantificación relativamente sencilla. La presencia de proteínas cerebrales en sangre y plasma o el aumento de IL-1β (Interleucina 1-beta) en líquido cefalorraquídeo (LCR) están asociados a la neuroinflamación, a la hiperexcitabilidad neuronal, a la reactividad glial, a la remodelación dendrítica y a la permeabilidad de la BHE que acompañan al proceso de epileptogénesis. En modelos animales y estudios clínicos se detectan microRNAs en plasma que se correlacionan con el proceso de epileptogénesis y presentan un perfil plasmático característicos que permiten diferenciar el tipo de crisis epiléptica (13).
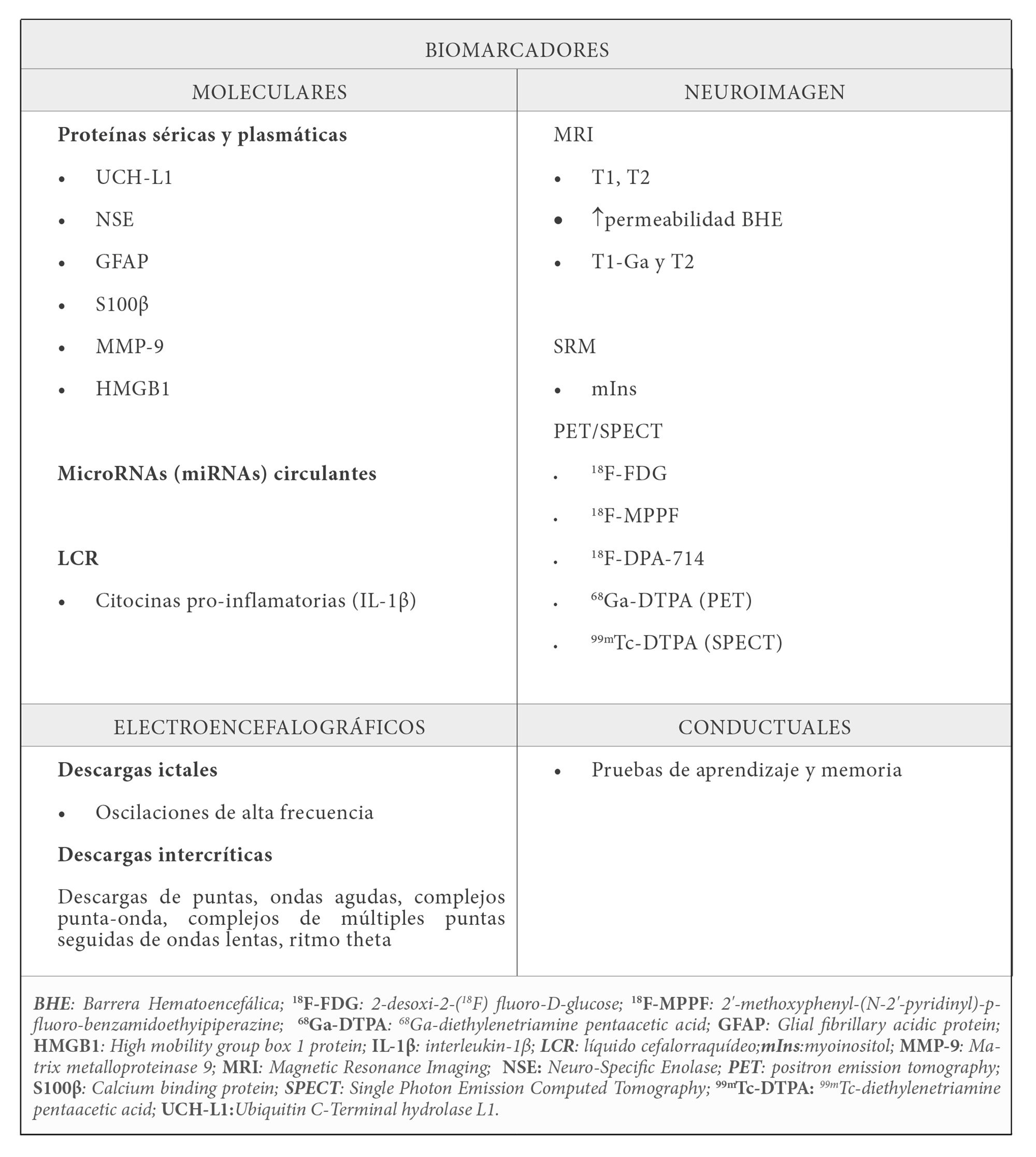
Los biomarcadores de imagen tienen una gran potencialidad por su traslación a la clínica. De hecho, las técnicas de PET, la tomografía computarizada por emisión de fotón único (SPECT, acrónimo inglés de Single Photon Emission Computerized Tomography) y la MRI se utilizan habitualmente en la clínica. Además, presentan una ventaja añadida y es que permiten realizar estudios longitudinales intra-individuo aportando información relevante sobre la evolución de la epileptogénesis.
Las técnicas de MRI utilizan secuencias de imagen estructural ponderada en T1 o T2 en la identificación del volumen hipocampal e intensidad de señal en regiones mesiales temporales y se han aplicado tanto en el ámbito clínico como en modelos animales de epilepsia (14). También permite evaluar cambios en la permeabilidad de la BHE con secuencias ponderadas en T1-Ga (Ga, gadolinio) y T2. La espectroscopia de resonancia magnética (MRS, acrónimo inglés de Magnetic Resonance Spectroscopy) detecta metabolitos cerebrales asociados con el SE como el N-acetil aspartato (NAA), los neurotransmisores GABA y glutamato, metabolitos como el lactato y biomarcadores de glía como el mio-inositol (mIns), cuyos niveles aumentan en fases iniciales de la epileptogénesis en varios modelos animales de SE (15).
Los estudios de PET con 2-desoxi-2-(18F) fluoro-D-glucosa (18F-FDG) en diferentes modelos animales de ELT detectan hipermetabolismo cerebral tras las crisis epilépticas agudas (16,17). Además, en el periodo silente es característico el hipometabolismo cerebral que es predictivo de la generación de crisis epilépticas en modelos de kindling químico (18). En las fases iniciales de la epileptogénesis también se han identificado alteraciones en la expresión de biomarcadores de microglía como la proteína translocadora de 18 kDa de peso molecular (TSPO) (19). Más aún, los datos preclínicos sugieren que la valoración de la TSPO mediante PET es útil cómo índice de la respuesta terapéutica o de la refractariedad a los tratamientos antiepilépticos (19, 20).
Los estudios de SPECT permiten evaluar el grado de permeabilidad de la BHE mediante el uso de trazadores como el 99mTc-DTPA. Trazadores similares con Ga (68Ga-DTPA) también se utilizan en la técnica PET con el mismo fin.
La técnica de EEG permite registrar los potenciales eléctricos generados por la actividad cerebral. Las descargas epilépticas ictales se manifiestan como oscilaciones de alta frecuencia y son biomarcadores de alta fiabilidad. El patrón de descargas obtenido durante los periodos intercríticos es biomarcador de confirmación y se caracteriza por la presencia de descargas de puntas, ondas agudas, complejos punta-onda y complejos de múltiples puntas seguidas de ondas lentas. En los modelos de ELT es frecuente registrar una actividad EEG theta.
Por último, comentar también un grupo de biomarcadores que explora los cambios conductuales o comportamentales asociados a la epileptogénesis. Pruebas de aprendizaje y memoria en el modelo animal de litio-pilocarpina evidencian una reducción de los índices de aprendizaje y de memoria con potencial valor predictivo de la epileptogénesis. Aunque la traslación de estos biomarcadores al ámbito clínico está aún por ser validada, su fácil adquisición y reducido coste los hacen un tipo de biomarcador prometedor (21).
5. HIPOMETABOLISMO CEREBRAL COMO BIOMARCADOR DE EPILEPTOGÉNESIS
En el cerebro, prácticamente toda la glucosa se oxida a CO2 y H2O a través de la glucólisis y la maquinaria respiratoria mitocondrial. La relación entre el consumo de O2 y la producción de CO2 en el cerebro es cercana a 1, de lo que se deduce que los carbohidratos, y la glucosa en particular, son los sustratos exclusivos del metabolismo oxidativo en el cerebro (22). La técnica de PET con un radiotrazador análogo de la glucosa (18F-FDG), es de las principales técnicas de imagen que permiten explorar las variaciones en el metabolismo energético cerebral. Así, cuando se administra 18F-FDG, las áreas con mayor captación de 18F-FDG corresponden con aquellas que tienen una mayor actividad funcional (23).
Teniendo en cuenta que las deficiencias en todas las principales vías metabólicas cerebrales son una característica de muchas enfermedades neurodegenerativas (24), es razonable que las alteraciones en el metabolismo cerebral puedan afectar la excitabilidad de la red neuronal, así como muchos otros aspectos que sustentan la epileptogénesis. En este contexto, los estudios de neuroimagen PET en pacientes con ELT muestran un aumento en el consumo de O2 y de glucosa en el periodo ictal. La energía necesaria para mantener la hiperactividad neuronal durante la crisis se obtiene a través de la vía aeróbica neuronal y del aumento de la glucólisis astrocitaria con un aumento de los niveles de la lactato deshidrogenasa (LDH). Sin embargo, la fase interictal se caracteriza por hipometabolismo en la zona epileptógena del lóbulo temporal.
Se ha sugerido que este hipometabolismo, asociado a un déficit energético, podría ser un reflejo de la pérdida neuronal, de la desaferenciación neuronal, de la reducción del flujo sanguíneo y/o podría ser considerado como un mecanismo adaptativo inicial del metabolismo celular que mantendría la homeostasis durante el proceso epileptogénico (25).
Este patrón de neuroimagen 18F-FDG-PET también se ha observado en modelos animales de epilepsia [Figura 1]. Así, en las primeras horas tras la inducción del SE se produce un aumento de captación de 18F-FDG que evoluciona en las 24-48 h posteriores hacia un marcado hipometabolismo cerebral (26). En el modelo animal de epilepsia inducido con PTZ, este hipometabolismo cerebral tiene un valor predictivo de la resistencia y/o de la vulnerabilidad a desarrollar crisis epilépticas (18). Más aún, en el modelo inducido por litio-pilocarpina se ha demostrado que el hipometabolismo se puede prevenir e incluso revertir mediante tratamientos farmacológicos con metirapona (20). Estos datos preclínicos junto con los obtenidos en estudios clínicos longitudinales en niños con epilepsia refractaria a tratamientos anticonvulsivantes (27) apuntan a que las alteraciones temporales del metabolismo cerebral de la glucosa no sólo son un biomarcador cuantificable asociado a las crisis epilépticas sino, aún más importante, son un índice de respuesta a las intervenciones terapéuticas efectivas.

6. ASTROCITOS EN LA REGULACIÓN DEL METABOLISMO CEREBRAL EN LA EPILEPTOGÉNESIS
El daño cerebral y la reactividad glial están estrechamente relacionados siendo dos de las características más comunes de la mayoría de las enfermedades neurodegenerativas, incluida la fase temprana de la epileptogénesis (28). En la ELT y otros síndromes epilépticos, la gliosis es una característica neuropatológica común y, lo que es más importante, a menudo se asocia con la resistencia al tratamiento farmacológico. La esclerosis hipocampal se caracteriza por gliosis reactiva, pérdida neuronal y cicatrización glial (29) estando asociada con deterioro cognitivo (30). Dichas alteraciones celulares (astrogliosis, neurodegeneración y muerte neuronal) están presentes en modelos animales de ELT como el de litio-pilocarpina (16, 17). En las áreas cerebrales afectadas, las células gliales, tanto microglía como astrocitos, experimentan cambios fisiológicos y moleculares que acompañan el desarrollo y la progresión de la epileptogénesis (31, 32).
Los astrocitos reactivos participan en: (i) la captación del glutamato potencialmente excitotóxico, (ii) la producción de glutatión, (ii) la liberación de adenosina, (iv) la facilitación de la reparación de la BHE, (v) la estabilización del líquido extracelular y del equilibrio electrolítico; (vi) la supresión de las células inflamatorias y de los agentes infecciosos. Estas funciones de la glía en las fases iniciales de la epileptogénesis está ganando atención y supone un nuevo abordaje al estudio de la epileptogénesis (33-36). En la fase tardía de la epileptogénesis, los cambios en las subunidades de los receptores GABA, AMPA y NMDA y la plasticidad sináptica se suman a los que ocurren durante la fase temprana, contribuyendo en conjunto a la hiperexcitabilidad típica que caracteriza a la enfermedad (37).
Los astrocitos parecen modular la actividad epiléptica. Las oscilaciones de las concentraciones intracelulares de Ca2+ en los astrocitos son probablemente buenos indicadores de cambios tempranos en la actividad glial en condiciones patológicas (38). Cambios en la activación de astrocitos y microglía se han observado en pacientes con epilepsia, así como en modelos experimentales de ELT (AK y pilocarpina) y en modelos de crisis agudas (4-AP) (17, 20). La reactividad astrocitaria, medida por cambios morfológicos y por el aumento de GFAP (acrónimo inglés de Glial Fibrillary Acidic Protein), se ha detectado en las primeras horas después del inicio del SE [Figura 2].
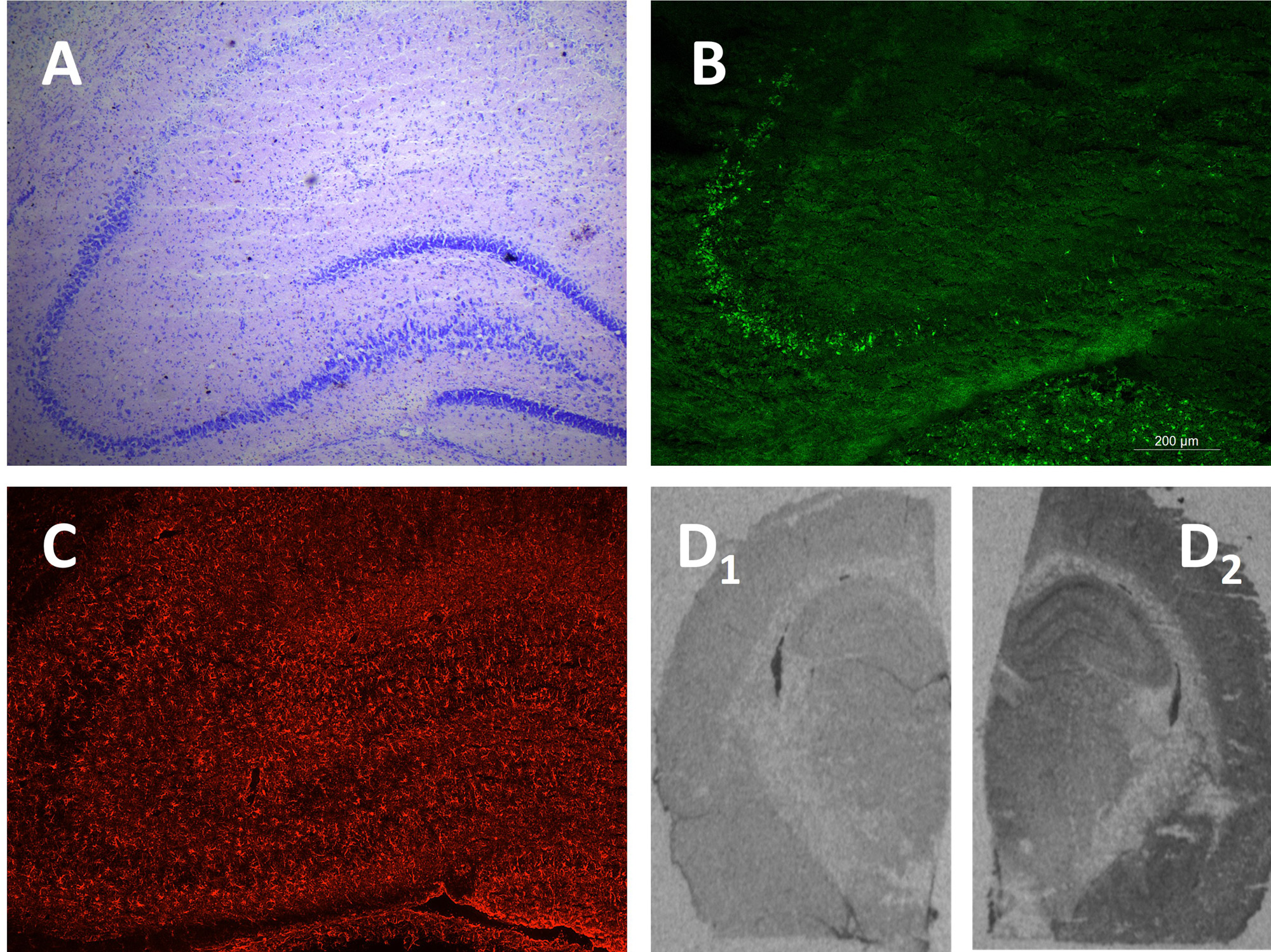
La participación de astrocitos reactivos en la modulación de las crisis epilépticas se ha relacionado con sus efectos reguladores de los niveles extracelulares de glutamato y K+ a través de diversos mecanismos que afectan a la excitabilidad neuronal y a la susceptibilidad a las crisis epilépticas (39). El aumento de la expresión de la TSPO [Figura 2] se considera un biomarcador de inflamación mediada por microglía (20). Además, como se mencionó anteriormente, los datos preclínicos sugieren que las mediciones de TSPO por PET podrían convertirse en una herramienta útil para evaluar la respuesta terapéutica o la resistencia a tratamientos antiepilépticos (19, 20, 40).
En conjunto, los cambios en la actividad astrocítica y microglial parecen contribuir a los mecanismos neuromoduladores subyacentes a las crisis epilépticas y, por lo tanto, se están considerando como nuevas dianas terapéuticas (39).
7. NUEVA APROXIMACIÓN AL ESTUDIO DE LA EPILEPTOGÉNESIS
Los astrocitos son uno de los tipos celulares más abundantes en el cerebro y su función es crucial regulando el metabolismo cerebral y estando involucrados en múltiples funciones relevantes para la homeostasis del sistema nervioso central (41).
Los astrocitos se localizan entre los vasos sanguíneos y las neuronas y tienen un papel específico en el metabolismo de la glucosa ya que son capaces de detectar y responder a las señales metabólicas (pO2, pCO2, pH) y de participar en la regulación del transporte de glucosa (42). La interrelación metabólica entre astrocitos y neuronas es esencial permitiendo la conversión de glucosa en lactato a través de la vía glucolítica (43).
El lactato se exporta a las neuronas, donde se convierte en piruvato para producir la molécula de energía de la célula ATP. Además, los astrocitos, a diferencia de las neuronas, presentan depósitos de glucógeno que puede ser movilizado ante un aumento en los requerimientos energéticos.
Un nuevo enfoque experimental para el estudio de la contribución de la reactividad glial en la epileptogénesis implica el uso de modelos murinos de quimiogenética. Las técnicas de quimiogenética se basan en la inserción de receptores diseñados para ser activados exclusivamente por fármacos de diseño (DREADDs, acrónimo inglés de Designer Receptors Exclusively Activated by Designer Drugs) en las células nerviosas. Hoy en día, se pueden desarrollar ratones que expresan receptores silenciosos en astrocitos (como por ejemplo el receptor muscarínico humano M3) que pueden ser activados selectivamente mediante la administración sistémica del fármaco agonista clozapina N-óxido (CNO) (44). Esta técnica se utiliza para modificar la actividad astrocitaria que en general resulta en cambios en la señalización del Ca2+ y diversos efectos en otras células cerebrales (45, 46).
La principal ventaja de esta técnica es la activación o inhibición específica de los astrocitos frente al enfoque farmacológico clásico que afecta de manera menos específica pudiendo alterar la función neuronal y/o producir diferentes efectos secundarios (47-50).
Como se ha comentado anteriormente, el proceso de epileptogénesis se caracteriza por un aumento de GFAP (biomarcador molecular de activación astrocitaria) así como de cambios metabólicos (neuroimagen 18F-FDG-PET). Estudios preliminares en nuestro laboratorio aplicando técnicas de quimiogenética en un modelo experimental de ELT inducido por la administración intrahipocampal de AK apoyan la contribución del papel regulador de los astrocitos en las alteraciones metabólicas cerebrales asociadas a la epileptogénesis.
8. CONCLUSIONES
Los modelos animales de epilepsia por SE, como los generados por la administración de AK o pilocarpina, han demostrado presentar características fenotípicas similares a las observadas en la ELT con esclerosis hipocampal en humanos. Tanto en humanos como en modelos animales se han identificado biomarcadores moleculares de activación glial y de neuroimagen metabólica por PET que son detectables ya desde la fase temprana de la epileptogénesis. Más aún, el hipometabolismo cerebral, medido por neuroimagen 18F-FDG-PET, que aparece tras el SE en el modelo animal de litio-pilocarpina puede ser prevenido y/o revertido por tratamiento farmacológico. En esta misma línea, la activación astrocitaria mediante tecnología DREADDs (quimiogenética) reduce el hipometabolismo en el modelo de ELT inducido por administración intrahipocampal de AK. En conjunto, estos resultados abren una nueva perspectiva al estudio de la epileptogénesis que contempla el efecto modulador de los astrocitos.
Finalmente, teniendo en cuenta el coste y el esfuerzo que supone el desarrollo de fármacos antiepilépticos y la necesidad de disponer de biomarcadores, el uso de modelos animales constituye una herramienta crucial. La disponibilidad de biomarcadores neuronales y astrocitarios de epileptogénesis permitirá aplicar una medicina personalizada en base a las características neuropatológicas, temporales y de respuesta farmacológica del paciente.
AGRADECIMIENTOS
Este trabajo ha sido financiado por el MICINN PID2019-106968RB-100
BIBLIOGRAFÍA
- Fisher RS, van Emde Boas W, Blume W, et al. Epileptic seizures and epilepsy: definitions proposed by the international league against epilepsy (ILAE) and the international bureau for epilepsy (IBE). Epilepsia. 2005; 46: 470.
- Baulac M, de Boer H, Elger C, et al. Epilepsy priorities in Europe: A report of the ILAE-IBE Epilepsy Advocacy Europe Task Force. Epilepsia. 2015; 56(11):1687.
- Engel Jr. J, Pitkänen A, Loeb JA, et al. Epilepsy biomarkers. Epilepsia. 2013, 54(Suppl. 4):61.
- Engel Jr. J, Pitkänen A. Biomarkers for epileptogenesis and its treatment. Neuropharmacology 2020; 167:107735.
- Laxer KD, Trinka E, Hirsch LJ, et al. The consequences of refractory epilepsy and its treatment. Epilepsy Behav. 2014; 37:59-70.
- Schmidt D. Drug treatment strategies for epilepsy revisited: starting early or late? One drug or several drugs? Epileptic Disord. 2016; 18(4): 356.
- Pitkänen A, Löscher W, Vezzani A, et al. Advances in the development of biomarkers for epilepsy. Lancet Neurol. 2016; 15(8):843.
- Becker AJ. Review: Animal models of acquired epilepsy: insights into mechanisms of human epileptogenesis. Neuropathol Appl Neurobiol. 2018; 44(1):112.
- Martín ED, Pozo MA Animal Models for the Development of New Neuropharmacological Therapeutics in the Status Epilepticus. Curr Neuropharmacol. 2006; 4(1): 33-40.
- Nirwan N, Vyas P, Vohora D. Animal models of status epilepticus and temporal lobe epilepsy: a narrative review. Rev Neurosci. 2018; 29(7):757.
- Nadler JV, Perry BW, Cotman CW. Intraventricular kainic acid preferentially destroys hippocampal pyramidal cells. Nature 1978; 271:676.
- Lévesque M, Avoli M. The kainic acid model of temporal lobe epilepsy. Neurosci Biobehav Rev. 2013; 37(10 Pt 2):2887.
- Enright N., Simonato M, Henshall DC. Discovery and validation of blood microRNAs as molecular biomarkers of epilepsy: ways to close current knowledge gaps. Epilepsy Open. 2018; 3: 427.
- Polli RS, Malheiros JM, Dos Santos R, et al. Changes in Hippocampal Volume are Correlated with Cell Loss but Not with Seizure Frequency in Two Chronic Models of Temporal Lobe Epilepsy. Front Neurol. 2014; 1;5:111.
- Vezzani A, Balosso S, Ravizza T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat Rev Neurol. 2019; 15: 459.
- García-García L, Shiha AA, Bascuñana P, et al. Serotonin depletion does not modify the short-term brain hypometabolism and hippocampal neurodegeneration induced by the lithium–pilocarpine model of status epilepticus in rats. Cell Mol Neurobiol. 2016; 36 (4): 513-9.
- García-García L, de la Rosa RF, Delgado M, et al. Metyrapone prevents acute glucose hypermetabolism and short-term brain damage induced by intrahippocampal administration of 4-aminopyridine in rats. Neurochem Int. 2018; 113: 92-106.
- Bascuñana, P, Javela, J, Delgado, M, et al. [18F]FDG PET Neuroimaging Predicts Pentylenetetrazole (PTZ) Kindling Outcome in Rats. Mol Imaging Biol. 2016; 18:733.
- Bascuñana P, García-García L, Javela J, et al. PET neuroimaging reveals serotonergic and metabolic dysfunctions in the hippocampal electrical kindling model of epileptogenesis. Neuroscience. 2019; 409: 101-10.
- García-García L, Shiha AA, Fernández de la Rosa R, et al. Metyrapone prevents brain damage induced by status epilepticus in the rat lithium-pilocarpine model. Neuropharmacology. 2017; 123:261.
- Simonato M, Agoston DV, Brooks-Kayal A, et al., Identification of clinically relevant biomarkers of epileptogenesis. A strategic roadmap. Nat Rev Neurol. 2021; 17(4): 231.
- Dienel GA. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol Rev. 2019; 99: 949-1045.
- Sokoloff L, Reivich M, Kennedy C, et al. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem. 1977; 28(5):897-916.
- Blass JP. Brain metabolism and brain disease: is metabolic deficiency the proximate cause of Alzheimer dementia?. J Neurosci Res. 2001; 66(5):851.
- Jupp B, Williams J, Binns D. Hypometabolism precedes limbic atrophy and spontaneous recurrent seizures in a rat model of TLE. Epilepsia. 2012; 53(7):1233.
- Reid AY, Staba RJ. Limbic Networks. Int Rev Neurobiol. 2014; 114:89-120.
- Govil-Dalela T, Kumar A, Behen ME, Harry T. Chugani HT, Juhász C. Evolution of lobar abnormalities of cerebral glucose metabolism in 41 children with drug-resistant epilepsy. Epilepsia. 2018; 59:1307.
- Henshall DC, Engel T. Contribution of apoptosis-associated signaling pathways to epileptogenesis: lessons from Bcl-2 family knockouts. Front Cell Neurosci. 2013; 7:110.
- Al Sufiani F, Ang LC. Neuropathology of temporal lobe epilepsy. Epilepsy Res Treat. 2012; 2012:624519.
- Rattka M , Brandt C, Löscher W. The intrahippocampal kainate model of temporal lobe epilepsy revisited: epileptogenesis, behavioral and cognitive alterations, pharmacological response, and hippocampal damage in epileptic rats. Epilepsy Res. 201; 103(2-3):135.
- Devinsky O, Vezzani A, Najjar S, De Lanerolle NC, Rogawski MA. Glia and epilepsy: excitability and inflammation. Trends Neurosci. 2013; 36(3):174.
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology Acta Neuropathol. 2010; 119(1):7-35.
- Horner PJ, Gage FH. Regenerating the damaged central nervous system. Nature. 2000; 407(6807):963.
- Fawcett JW, Asher RA. The glial scar and central nervous system repair. Brain Res Bull. 1999; 49(6):377.
- Giulian D. Reactive glia as rivals in regulating neuronal survival. Glia. 1993; 7(1):102.
- Li FQ, Wang T, Pei Z, Liu B, Hong JS. Inhibition of microglial activation by the herbal flavonoid baicalein attenuates inflammation-mediated degeneration of dopaminergic neurons. J Neural Transm. (Vienna). 2005; 112(3):331.
- Jabs R, Seifert G, Steinhäuser C. Astrocytic function and its alteration in the epileptic brain. Epilepsia. 2008; 49 Suppl 2:3-12.
- Ujita S, Sasaki T, Asada A et al. cAMP-dependent calcium oscillations of astrocytes: An implication for pathology. Cereb Cortex. 2017; 27(2):1602.
- Mahmoud S, Gharagozloo M, Simard C, Gris D. Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cells. 2019; 8(2):184.
- Bar‐Klein G, Klee R. Brandt C et al. Isoflurane prevents acquired epilepsy in rat models of temporal lobe epilepsy. Ann Neurol. 2016, 80(6), 896-908.
- Allen NJ, Barres BA. Glia-more than just brain glue. Nature 2009; 457: 675.
- Marina N, Turovsky E, Christie IN et al. Brain metabolic sensing and metabolic signaling at the level of an astrocyte. Glia. 2018; 66:1185.
- Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011; 14(6):724.
- Armbruster BN, Li X, Pausch MH, Herlitze S, Roth BL. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc Natl Acad Sci USA. 2007; 104 (12) 5163.
- Bonder DE, McCarthy KD. Astrocytic Gq-GPCR-Linked IP3R-Dependent Ca2+ Signaling Does Not Mediate Neurovascular Coupling in Mouse Visual Cortex In Vivo. J Neurosci. 2014; 34 (39): 13139.
- Shen W, Chen S, Liu Y, Han P, Ma T, Zeng LH. Chemogenetic manipulation of astrocytic activity: Is it possible to reveal the roles of astrocytes? Biochem Pharmacol. 2021; 186:114457.
- Jones ME, Paniccia JE, Lebonville CL, Reissner KJ & Lysle DT. Chemogenetic Manipulation of Dorsal Hippocampal Astrocytes Protects Against the Development of Stress-enhanced Fear Learning. Neuroscience. 2018; 388:45-56.
- Lim EY, Ye L & Paukert M. Potential and Realized Impact of Astroglia Ca2+ Dynamics on Circuit Function and Behavior. Front. Cell. Neurosci. 2021; 15:682888.
- MacDonald AJ, Holmes FE, Beall C, Pickering AE, Ellacott KLJ. Regulation of food intake by astrocytes in the brainstem dorsal vagal complex. Glia 2020; 68(6):1241.
- Nwachukwu KN, Evans WA, Sides TR, Trevisani CP, Davis A, Marshall SA. Chemogenetic manipulation of astrocytic signaling in the basolateral amygdala reduces binge-like alcohol consumption in male mice. J Neurosci Res. 2021; 99:1957.
DECLARACIÓN DE TRANSPARENCIA
Los autores/as de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
ranm tv
Miguel A. Pozo
Paseo Juan XXIII, 1· 28040 Madrid
Tlf.: +34 91 394 32 94 | E-Mail: pozo@ucm.es
Año 2022 · número 139 (02) · páginas 140 a 149
Enviado*: 03.05.22
Revisado: 12.05.22
Aceptado: 18.05.22
* Fecha de lectura en la RANM