

Resumen
La enfermedad de Alzheimer (EA) es una compleja patología neurodegenerativa que afecta principalmente a la población de edad avanzada. Hasta la fecha, las terapias dirigidas a modificar el curso de la enfermedad no han resultado efectivas, siendo necesario descifrar en mayor detalle los procesos patogénicos que acontecen en la EA. Estudios recientes indican que la EA y otras enfermedades neurodegenerativas comparten la acumulación de proteínas con plegamiento alternativo, las cuales pueden actuar como agentes infecciosos y propagarse por el cerebro afectando al plegamiento nativo de otras proteínas. El péptido amiloide (Aβ), característico de la EA, puede adoptar una conformación de hoja β-plegada, adquiriendo ese potencial de agregar y generar intermediarios mal plegados que pueden actuar como “semillas” propagando la enfermedad dentro del cerebro. Estos estudios sugieren que dichas semillas son distintas dependiendo del paciente y subtipo de EA, albergando diferente capacidad de propagarse y toxicidad. Para poder conocer qué especies de Aβ o isoformas del péptido son más tóxicas y tienen mayor potencial de agregación, se han inoculado extractos cerebrales de pacientes de EA en modelos murinos transgénicos para esta patología. Estos estudios han proporcionado mucha información, sin embargo, la mayoría de los modelos empleados contienen una o más mutaciones dominantes para la EA familiar. Esto es de gran valor para estudiar las formas familiares de la enfermedad, pero no los casos de inicio tardío (LOAD), que representan la mayoría. En consecuencia, es necesario el desarrollo de nuevos modelos animales que representen el LOAD y permitan su estudio. En este aspecto, un nuevo modelo humanizado (hAβ-KI) generado en el centro UCI-MODEL-AD de California, recapitula ciertos rasgos del LOAD, suponiendo un modelo viable para entender cómo Aβ se propaga y favorece la progresión de la patología en el cerebro, permitiendo el desarrollo de nuevas estrategias terapéuticas para la EA.
Abstract
Alzheimer´s disease (AD) is a complex neurodegenerative disorder that affects mainly the elder population. To date, modifying therapies have not proven to be successful, and therefore, a major effort is necessary to unravel the pathogenic processes underlying AD. New evidences indicate that AD and many other neurodegenerative diseases have commonly the accumulation of misfolded proteins that have the potential to act as infectious agents, and propagate through the entire brain, affecting native proteins. The amyloid peptide (Aβ), characteristic of AD, can adopt a β-sheet conformation acquiring the potential to aggregate and generate misfolded intermediates that can act as ‘’seeds’’ to propagate disease in the brain. Recent findings suggest that these seeds are structurally different depending on the patients and AD subtype, showing different propagation capacity and toxicity. In order to find out which Aβ species or isoforms are more toxic and have greater potential to aggregate, brain extracts from AD patients have been inoculated into transgenic AD mice brains. These approaches have provided much information, however, most of the models contain one or multiple dominant mutations of familial AD cases. This is of great value to mimic the familial forms of the disease, but not the late onset AD (LOAD), which constitute the majority of the cases. Therefore, new animal models are urgently necessary to circumvent these limitations and properly dissect the pathogenic mechanisms of this common form of the disease. As such, a new humanized Aβ model, called hAβ-KI, that has been generated by UCI-MODEL AD center at California, recapitulates certain features of LOAD. This might represent a useful LOAD model to understand how Aβ spreads and favors the progression of the pathology in the brain, hopefully enabling the development of effective therapeutic interventions for AD.
Palabras clave: Alzheimer; Amiloide; Propagación; Modelos animales.
Keywords: Alzheimer; Amyloid; Propagation; Animal models.
INTRODUCCIÓN
La enfermedad de Alzheimer (EA) es una compleja patología neurodegenerativa y la forma más común de demencia (1). Actualmente, en el mundo existen unos 50 millones de individuos que sufren la enfermedad, una cifra que se estima continuará creciendo hasta sobrepasar los 100 millones de personas en 2050 (2). Mientras que las muertes causadas por otras enfermedades, tales como las cardiovasculares, cáncer o SIDA han disminuido en los últimos años, las asocidadas con la EA han aumentado un 68% durante la última década (3). El incremento significativo del número de casos está generando un elevado impacto a nivel sociosanitario y económico, y a pesar de que recientemente la Agencia del Medicamento (FDA) de Estados Unidos ha autorizado el uso terapéutico de dos anticuerpos monoclonales, aducanumab (4) y lecanemab (5), su seguridad y eficacia clínica aún necesita ser confirmada. La EA fue inicialmente descrita a nivel neuropatológico al inicio de 1900 por Alois Alzheimer y, a día de hoy, se ha avanzado de forma significativa en el conocimiento celular y molecular de esta enfermedad (1–3). Por ejemplo, se conoce que las lesiones características que presentan los cerebros de los pacientes, denominadas placas seniles y ovillos neurofibrilares, están formadas por agregados fibrilares de las proteínas β-amiloide y tauhiperfosforilada, respectivamente. Sin embargo, la formación de estos agregados y el mecanismo por el cual se propagan de una región a otra del cerebro aún no se ha determinado. En relación a esto, nuevas evidencias científicas sugieren que ambos péptidos con plegamiento alternativo tienen una alta capacidad de propagarse y afectar a proteínas nativas para que adquieran una conformación aberrante, favoreciendo la progresión de la enfermedad. Por tanto, estos estudios sugieren que dichas proteínas adquieren el mismo patrón acontecido en las enfermedades priónicas, siendo capaces de infectar distintas regiones cerebrales (6,7). Una mayor comprensión de estos mecanismos patogénicos es de gran importancia para el desarrollo de nuevas terapias efectivas que puedan detener o retrasar el desarrollo de esta enfermedad, siendo determinantes para mejorar la calidad de vida de los pacientes que sufren la EA.
1. Características Histopatológicas de la Efermedad de Alzheimer
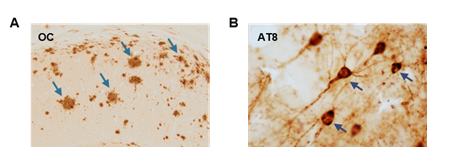
A nivel molecular, la EA se define como una doble proteinopatía caracterizada por la acumulación a nivel extracelular del péptido β-amiloide o Abeta (Aβ) formando las llamadas placas seniles o amiloides, y por la acumulación intracelular de la proteína tau hiperfosforilada en estructuras conocidas como ovillos neurofibrilares (3,8,9) (Figura 1). Ambas características histopatológicas resultan de la agregación de proteínas con un plegamiento alternativo generándose conformaciones neurotóxicas con capacidad de propagación (6,10).
1.1. Patología β-amiloide
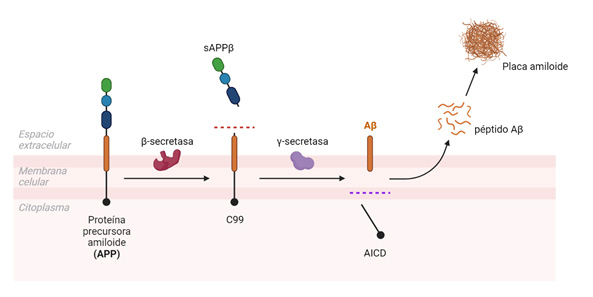
Las placas amiloides son estructuras insolubles que se originan por la acumulación progresiva del péptido Aβ liberado al parénquima cerebral (Figura 1A), que a su vez deriva del procesamiento proteolítico de la proteína precursora amiloide (APP) por las enzimas β- y γ-secretasas (11,12,13) (Figura 2). Esta vía amiloidogénica se encuentra exacerbada en pacientes de Alzheimer, y ciertas mutaciones en la secuencia de APP, o de las presenilinas 1 (PS1) o 2 (PS2) que constituyen la subunidad catalítica de la γ-secretasa, promueven la formación de formas más hidrofóbicas y tóxicas del Aβ, acelerando el inicio de la patología.
Precisamente, estas mutaciones se heredan con carácter autosómico dominante y son las responsables de las formas familiares de la EA (14). El Alzheimer familiar, también referido como Alzheimer de inicio temprano, se manifiesta a edades tempranas en los pacientes, en concreto, en torno a los 40 años de edad. Sin embargo, se da en tan solo un 1% de los casos totales de Alzheimer, siendo más prevalente (99% de los casos restantes) la forma esporádica de la EA o Alzheimer de inicio tardío. En ésta, como su nombre indica, la manifestación clínica es más tardía y su origen se ve influenciado por una serie de factores que actúan de forma sinérgica, donde algunos de los más importantes son: el envejecimiento, factores de riesgo genético (APOE4, TREM2, y otras variantes identificadas por estudios de asociación del genoma completo (GWAs en inglés), enfermedades comórbidas presentes en los pacientes y factores ambientales (14,15,16). No obstante, los mecanismos moleculares y celulares que subyacen a este tipo de EA, se encuentran ante una intensa investigación, y a día de hoy, sigue siendo un enigma por resolver.
La longitud del péptido β-amiloide generado puede variar entre los 36 y 43 aminoácidos dependiendo del sitio de corte de la γ-secretasa, siendo los péptidos de 40 (Aβ40) y 42 aminoácidos (Aβ42) los más frecuentemente encontrados en pacientes con Alzheimer (11,17). La isoforma Aβ42 es más propensa a agregar y formar placas insolubles en el parénquima, mientras que el péptido Aβ40 se encuentra mayoritariamente formando depósitos a nivel vascular, dando lugar a lo que se conoce como angiopatía cerebral amiloide o CAA (11).
Adicionalmente, se han encontrado más isoformas del péptido amiloide originadas tras sufrir diversas modificaciones postraduccionales, incluyendo Aβ piroglutamato truncado (AβN3pE) o Aβ fosforilado en la serina 8 (pSer8Aβ) (11,15). Todas estas isoformas de Aβ están asociadas con la progresión de la patología clínica de la EA, y se sugiere que participan en diferentes fases de la formación de placas y maduración de la enfermedad (11).
Numerosos estudios han indicado que distintas especies del péptido Aβ son capaces de generar agregados de diversos tipos, que a su vez inducen una toxicidad variable en los cerebros de los pacientes (11,17,18,19). Por tanto, cada individuo puede presentar una patología que difiere de los demás, incrementando la dificultad de estudiar, diagnosticar y tratar la EA.
Todo esto resalta la alta importancia de conocer qué isoformas son las más dañinas, citotóxicas, o las que tienen una mayor capacidad de agregar, entre otros aspectos. Su caracterización es esencial para poder desarrollar nuevas dianas terapéuticas dirigidas a modificar la progresión de la patología amiloide, capaces de prevenir la formación de las isoformas citotóxicas del Aβ.
1.1.1 Tipos de placas amiloides
Desde que se describió por primera vez la EA, se ha registrado la existencia de depósitos amiloides de distinta naturaleza en el cerebro de los pacientes, haciendo que cada individuo presente unos rasgos histopatológicos que difieren levemente entre ellos.
En primer lugar, los depósitos de Aβ en el parénquima cerebral son lo que se conoce como placas seniles, mientras que cuando se acumulan en los vasos sanguíneos del cerebro se denomina CAA (11). Dentro de la categoría de placas se encuentran las fibrilares, caracterizadas por la presencia del péptido Aβ en una conformación de hoja β-plegada (también denominado amiloide), además de ser positivas a los colorantes rojo Congo y Tioflavina-S, y correlacionar con la manifestación clínica de la demencia. Por otro lado, las placas pueden ser no fibrilares, comúnmente llamadas placas difusas. Éstas, por el contrario, son negativas a dichos colorantes y están más presentes en individuos sin síntomas de declive cognitivo.
Las placas amiloides también pueden clasificarse según la presencia o ausencia de componente neurítico. De esta forma, una placa será neurítica cuando presente a su alrededor axones y sinapsis dilatadas llenas de vesículas autofágicas (neuritas distróficas). Las placas neuríticas además presentan microglía y astroglía reactiva asociada. Adicionalmente, las placas fibrilares se pueden subdividir en placas clásicas con un núcleo central compacto de amiloide rodeado de una corona de Aβ oligomérico no fibrilar (classic-cored plaques en inglés), o bien en placas primitivas, que, por el contrario, no poseen un núcleo denso de amiloide (11,20,21).
Todas estas son las variantes de placas más comunes, aunque recientemente se han descrito nuevos tipos de depósitos. Por ejemplo, en los pacientes que sufren mutaciones familiares en el gen PSEN1, se ha observado un tipo de placa con aspecto de ovillo de lana (cotton wool plaques, en inglés) (11,15,21).
Por otro lado, también se ha encontrado en pacientes de Alzheimer de inicio temprano con elevada patología vascular y un intenso componente neuroinflamatorio, las denominadas placas de granulado grueso (coarse-grained plaques, en inglés) (11).
Toda esta amplia gama de depósitos amiloides refleja la heterogeneidad patológica entre los pacientes, haciendo que sea más difícil la búsqueda de un tratamiento que funcione en todos los individuos, puesto que cada uno presenta un subtipo de patología.
Conocer el proceso que subyace la formación de placas y qué papel tiene el péptido Aβ en la amiloidogénesis, son aspectos clave para poder frenar el avance de la patología.
1.2. Patología tau
De forma complementaria, y en sinergia con la patología amiloide, se encuentra la patología tau, el otro rasgo neuropatológico de la EA. Consiste en la acumulación intraneuronal de la proteína tau hiperfosforilada en estructuras conocidas como ovillos neurofibrilares (NFTs) (22) (Figura 1B). Fisiológicamente, tau es una proteína asociada a microtúbulos expresada abundantemente en el cerebro a nivel axonal. Su función se centra en promover el ensamblaje de los microtúbulos axonales mediante su unión a la tubulina. Sin embargo, bajo condiciones patológicas, tau se fosforila de forma exacerbada y anómala, pierde su unión a tubulina y se trasloca del axón a los compartimentos somatodendríticos, agregándose para formar NFTs (23). Esto conlleva la desestabilización de los microtúbulos y fallos en el transporte axonal, comprometiendo la supervivencia neuronal. Se piensa que la patología tau es una de las principales causas de la muerte neuronal en la EA, y su aparición se correlaciona con el declive cognitivo de los individuos (24). Sin embargo, aparece en fases avanzadas de la enfermedad, cuando el depósito de Aβ extracelular ya ha alcanzado altos niveles.
1.3. Hipótesis de la cascada amiloide
La interacción entre ambas proteinopatías, tau y amiloide, se explica con la hipótesis de la cascada amiloide postulada por Hardy y Higgins en 1992 (25), la cual mantiene que el proceso de acumulación y agregación del péptido Aβ es clave en el inicio y propagación de la enfermedad, dado que la patología amiloide puede comenzar hasta 20 años antes de la manifestación clínica de la EA y de la acumulación de la proteína tau hiperfosforilada (1,15) . No obstante, el continuo fracaso de los ensayos clínicos cuyas dianas están dirigidas al péptido Aβ, ha llevado a cuestionar la solidez de esta hipótesis. Sin embargo, y a pesar de las controversias generadas, a día de hoy sigue vigente, aunque ha sufrido algunas modificaciones a lo largo de los últimos años como la incorporación de formas oligoméricas de Aβ (15,20). Aunque sean desalentadores, estos ensayos fallidos pueden no ser motivo suficiente para rechazar o disminuir el papel de Aβ en la patogénesis de la EA, debido a los complicados factores en torno al diseño y análisis de dichos estudios clínicos.
A pesar de que el conocimiento de esta enfermedad ha aumentado mucho en las últimas décadas, todavía no se conoce bien cómo la acumulación de Aβ es capaz de inducir el desarrollo de la patología tau y los procesos neurodegenerativos asociados en los pacientes con EA. Se piensa que para que esto ocurra, es necesaria la presencia de más factores además de la propia acumulación de amiloide. En este sentido, el componente genético es de gran importancia, pues se sabe que variantes genéticas en los genes APOE o TREM2 pueden tener un gran impacto sobre la propagación de la patología (1).
Por tanto, se plantea que el péptido amiloide, mediante una vía directa o indirecta, está afectando a la fisiología del cerebro, haciendo que se desencadenen la patología tau, la activación de células gliales, con el consecuente proceso neuroinflamatorio, y las manifestaciones clínicas que se observan. Además, la dotación genética de cada individuo hará que sea más o menos vulnerable a este proceso, o incluso resiliente al deterioro cognitivo. Dada su importancia, conocer cómo comienza y progresa la patología amiloide es fundamental, al igual que conocer qué factores de riesgo influyen en el proceso, para poder entender la EA y diseñar nuevas dianas terapéuticas.
2. Propagación de la patología mediante un mecanismo priónico
En la EA, al igual que en otras patologías neurodegenerativas, los agregados proteicos patogénicos pueden propagarse entre las diferentes regiones cerebrales a medida que avanza la enfermedad. Este tipo de trastornos se engloban dentro de las enfermedades asociadas a péptidos mal plegados o, como se denomina en inglés, protein misfolding diseases (PMDs) (26).
En este sentido, estudios liderados por los laboratorios de los doctores Soto y Jucker (6,19,27,28,29,30,31) han demostrado que una vez se inicia la patología amiloide, ésta es capaz de progresar y propagarse mediante un mecanismo priónico denominado “siembra” (seeding en inglés). Esto quiere decir que, formas mal plegadas, intermedias y oligoméricas de beta-amiloide (a las que se refiere como semillas o seeds) pueden actuar sobre monómeros nativos del péptido Aβ e inducir un cambio conformacional en ellos (10,32) (Figura 3). De esta manera, son reclutados y comienzan a agregar, formando estructuras oligoméricas y fibrilares que contribuyen a la propagación de la patología en el cerebro (9). Conocer cómo se produce el mecanismo de seeding y cuáles son los factores determinantes en este proceso, es clave para entender cómo se propaga en el cerebro la patología en la EA.
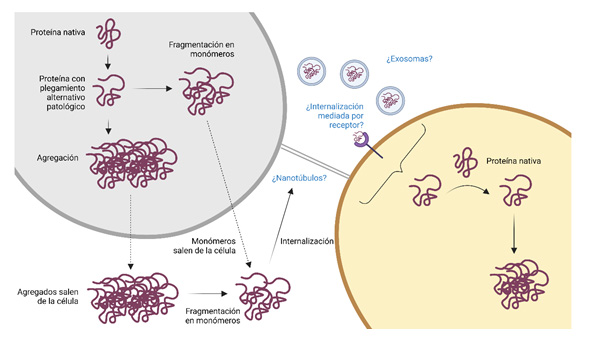
Como se ha mencionado antes, la EA puede presentarse de formas muy heterogéneas a nivel patológico y clínico, indicando que pueden existir diferentes subtipos de EA (33). En otras enfermedades priónicas, como la de Creutzfeldt-Jakob (CJD), la variabilidad reside en la presencia de distintas cepas de agregados proteicos en el cerebro. A diferencia de los virus o bacterias, donde las nuevas cepas surgen debido a mutaciones en las secuencias génicas, en el caso de los agregados de proteínas se piensa que las cepas representan variaciones estructurales en la conformación de esos péptidos (15). Al igual que ocurre en CJD y otras enfermedades priónicas animales, la presencia de diferentes cepas de priones en el cerebro es lo que determina el fenotipo clínico y patológico del paciente (34). Por este motivo, dado que, tanto Aβ como fosfo-tau han mostrado tener propiedades priónicas, se piensa que la heterogeneidad que se observa en los pacientes puede explicarse por la presencia de distintas cepas de agregados proteicos en sus cerebros (15).
Ahora bien, se pueden definir las cepas amiloides como proteínas con una estructura química idéntica, pero conformacionalmente distintas debido a que han adquirido un plegamiento alternativo como consecuencia de un agente externo (15,35). Cada una de estas especies tiene sus propias características bioquímicas y está asociada con diferente fenotipo clínico (34). A día de hoy, qué cepas de Aβ son las más tóxicas todavía se desconoce, aunque los estudios apuntan a que los agregados más pequeños y solubles correlacionan mejor con la progresión de la enfermedad, por lo que se piensan que son más perjudiciales que los agregados de mayor tamaño (15).
De forma adicional a este tipo de propagación, se encuentra lo que se denomina siembra heteróloga (cross-seeding en inglés o seeding heterólogo). Ocurre cuando oligómeros formados por una proteína mal plegada pueden promover el plegamiento alternativo de otra proteína diferente (36). Esto sucede en multitud de proteínas que se pliegan con una conformación de hoja β-plegada, denominadas comúnmente como amiloides funcionales. Este tipo de agregación podría explicar la diversidad de agregados que se encuentran en los pacientes con EA y, por ejemplo, explicaría por qué existen pacientes con más de una PMD, o por qué sufrir una PMD aumenta el riesgo de sufrir una segunda PMD. Todo esto añade dificultad y complejidad al estudio de la enfermedad, y son factores a tener en cuenta a la hora de desarrollar una terapia efectiva.
2.1. Evidencia científica a favor de la propagación priónica de la patología amiloide
La agregación y acumulación de péptidos de Aβ se consideran dos eventos clave en la patogénesis de la EA, y en esta revisión nos vamos a centrar en comentar estudios que nos han permitido avanzar en el conocimiento de este proceso patogénico. En relación a lo comentado, diversas evidencias científicas indican que la inoculación intracerebral de homogenados de cerebro (que contienen agregados de Aβ) en modelos murinos transgénicos, acelera la formación de depósitos de Aβ en los individuos inyectados, sugiriendo que los agregados de Aβ son capaces de propagarse por sí mismos por el cerebro de una forma similar a los priones (6,10,37). En este sentido, un estudio reciente (38) indica que los agregados de Aβ pueden circular por la sangre y penetrar en el cerebro, donde inducen el desarrollo de patología amiloide y déficits neuronales. En este estudio, un ratón con patología de la EA se encontraba en parabiosis con un ratón sano, y tras un tiempo, el sano desarrolló patología amiloide por estar en contacto con sangre del ratón que presentaba esta misma patología. En concordancia con este estudio, se ha demostrado que la infusión de sangre de animal transgénico de la EA en ratones modelo de la enfermedad, es capaz de inducir en estos animales receptores una aceleración de la patología amiloide (28).
Por otro lado, otros estudios interesantes muestran que la patología de Aβ no solo puede transmitirse entre primates no humanos (39), sino también entre humanos (19,40). En estos estudios se descubrió que los pacientes con deficiencia de hormona del crecimiento (GH) que habían sido tratados con esta hormona derivada de hipófisis humana postmortem, padecieron en su mayoría la enfermedad priónica Creutzfeldt-Jakob. No solo eso, sino que además dichos pacientes tratados con GH humana presentaban una mayor acumulación de Aβ en sus cerebros y vasos sanguíneos. Estos resultados concuerdan con la hipótesis de que el péptido amiloide estaba presente en el material humano usado en el tratamiento para la deficiencia en esta hormona. De ese modo, el Aβ, mediante un mecanismo de seeding, había inducido en un 50% de los pacientes una neuropatología similar a la de la EA.
En conjunto, estos estudios descritos anteriormente sugieren que las semillas de Aβ son claves en los procesos de propagación de la patología por todo el cerebro. Por tanto, es esencial realizar ensayos de agregación in vivo e in vitro que permitan conocer más profundamente cómo ocurre este fenómeno y qué tipo de semillas son las que participan de forma más activa en estos procesos patogénicos de propagación.
En la actualidad, podemos encontrar una amplia diversidad de estudios in vivo en los que los inóculos ricos en agregados amiloides presentan una naturaleza muy diversa. Por un lado, se pueden inyectar homogenados de cerebro, que provienen tanto de pacientes de Alzheimer, como de ratón transgénico (17). Por otro lado, pueden inyectarse preparados de Aβ sintético (37). En general, los resultados de dichos estudios indican que los inóculos sintéticos tienen una menor capacidad de propagar la patología amiloide comparados con los inóculos procedentes de modelos murinos o humanos. No obstante, los resultados de estas investigaciones en modelos murinos de la EA sugieren que i) los tipos de semillas presentan una configuración diferente, teniendo una mayor capacidad infectiva los extractos procedentes de muestras de humanos y ratón en comparación con extractos sintéticos, ii) en gran cantidad de pacientes de la EA la patología es mixta, pudiendo diferentes proteínas heterólogas participar en los procesos de agregación, iii) los extractos de humanos y modelos murinos contienen otros elementos adicionales que pueden participan en la propagación, tales como pueden ser las chaperonas (29,37). Esto demuestra el carácter priónico del péptido Aβ, la posibilidad de poder propagar la patología por sí mismo y la gran diversidad de posibles agregados con distinta capacidad infectiva tanto en los extractos cerebrales de pacientes de EA, como en los modelos murinos. Por tanto, un mayor conocimiento en estos fenómenos de seeding del péptido Aβ permitirá que este proceso represente una importante diana terapéutica para la EA, con el objetivo de mitigar o ralentizar el curso de la enfermedad.
2.2. Limitaciones y factores que influyen en los procesos de propagación de la patología amiloide
Tras considerar todos estos estudios, se puede concluir que el mecanismo de seeding se encuentra influenciado principalmente por cuatro parámetros: el tipo de semilla inyectado, el huésped que la recibe, la ruta de administración empleada y el tiempo de incubación de la semilla en el individuo (10) (Figura 4).
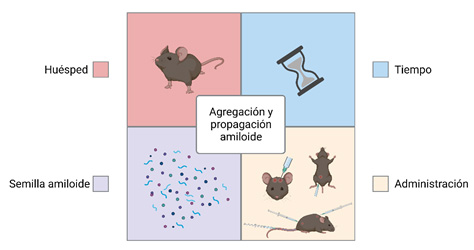
Este último, el tiempo de incubación, es altamente importante. Diversos estudios sugieren que no es la edad del animal inyectado lo que afecta a la propagación, sino el tiempo de incubación con las semillas de Aβ lo que realmente potencia la deposición de amiloide y su propagación a otras regiones del cerebro (17,31).
Adicionalmente, existen otros factores que influyen de forma significativa en la propagación de amiloide. Por ejemplo, la vía de administración de las semillas de Aβ es un nuevo aspecto a tener en cuenta. Se ha observado que la administración intraperitoneal de extractos ricos en Aβ pueden inducir amiloidosis en modelos murinos transgénicos para la enfermedad, aunque la concentración y el tiempo de incubación requeridos para ello es mayor que cuando la inoculación es intracerebral (19,30). De la misma forma, la administración oral de altas cantidades de extractos cerebrales de pacientes de Alzheimer y de ratones transgénicos envejecidos, no ocasiona un efecto en la patología cerebral (19).
Aunque los estudios anteriormente comentados han demostrado que tanto el tiempo de incubación como la ruta de administración son factores críticos en el tipo de agregado que se forma a nivel cerebral, es importante destacar que la mayoría de ellos se han realizado en modelos murinos que representan la forma de la enfermedad de inicio temprano. Si bien nos han ayudado a entender las bases generales de este trastorno neurodegenerativo, la mayoría de los casos asociados a la EA son de inicio tardío, por lo que es de gran importancia el desarrollo de nuevos modelos que reproduzcan esta forma mayoritaria de la EA, y nos permitan entender cómo la propagación de la patología amiloide ocurre en ellos.
Con ese objetivo, el equipo de la Universidad de California, Irvine (UCI), que forma parte del consorcio MODEL-AD (del inglés Model Organisms Development and Evaluation for Late-Onset Alzheimer’s Disease), liderado por el doctor Frank LaFerla, y en colaboración con nuestro grupo de investigación, ha conseguido desarrollar un nuevo modelo animal para la EA de inicio tardío, llamado hAb-KI, en el que se ha humanizado la secuencia del Aβ murino (41). Concretamente, se ha insertado la secuencia wild-type del Aβ humano en el locus del gen App de ratón, por lo que, al estar bajo el control de su promotor endógeno, se expresa de forma fisiológica y sin ninguna mutación. Este modelo mejora a sus antecesores amiloidogénicos, ya que no contiene mutaciones de la forma familiar, no se sobreexpresa el gen humano, no se encuentra modulado por promotores exógenos y el fragmento humanizado no está insertado de forma aleatoria en el genoma del roedor (42). Este nuevo modelo se caracteriza por presentar daños cognitivos, plasticidad sináptica, cambios en el volumen cerebral, alteraciones inflamatorias, gránulos positivos al ácido periódico de Schiff (PAS) asociados con la edad y cambios de expresión génica (Figura 5). Además, el exón 14 que codifica para la secuencia de Aβ de humano, se encuentra flanqueado por sitios loxP, permitiendo una supresión inducible de la expresión del péptido Aβ (41). Este modelo murino, donde se ha humanizado la secuencia de Aβ, puede servir como plataforma para introducir otros factores genéticos y ambientales asociados a la forma tardía del Alzheimer, y estudiar en más profundidad cómo estos factores pueden influir en el desarrollo y progresión de la EA, incluidos los agregados amiloideos.
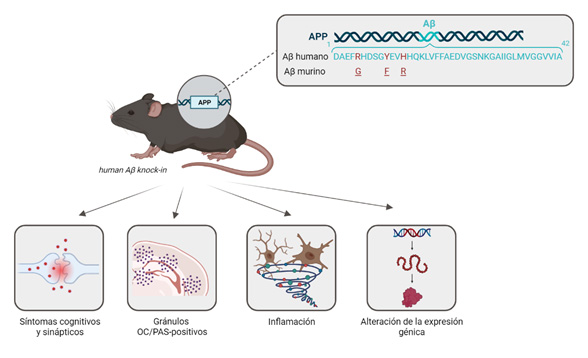
A pesar de que este modelo transgénico para LOAD es una herramienta prometedora, todavía existen ciertas limitaciones a considerar. Por ejemplo, el Aβ de humano expresado en estos ratones knock-in no consigue formar placas amiloides, aunque según los estudios in vitro, sus semillas de Aβ sí que son competentes para agregar (41). Los investigadores plantean que esto puede ser debido a que hace falta la influencia de otros factores de riesgo adicionales, o más tiempo, que permitan la formación de los agregados insolubles amiloides. A su vez, la introducción de otros genes humanizados, tales como los que codifican para las proteínas tau o ApoE, puede ser de vital importancia para comprender mejor la interacción entre los mismos, y generar mejores modelos que reproduzcan de forma más eficiente los mecanismos que subyacen en la forma tardía de la enfermedad.
CONCLUSIONES
A pesar de que el conocimiento sobre la patología celular y molecular de la EA no ha hecho más que crecer en las últimas décadas, aún no existe una terapia eficaz capaz de frenar el progreso de la enfermedad. Por tanto, continuar y aumentar los estudios y la investigación en este campo es de vital importancia. Si se consigue conocer en detalle cómo se inicia la patología o cuáles son las cepas que más influyen en la propagación y en la formación de agregados proteicos más citotóxicos, entre otros aspectos, se podrán desarrollar terapias más avanzadas que nos permitan, en cierto grado, prevenir la EA. Para ello, es de una alta relevancia biológica hacer estudios de seeding, que proporcionen información de cómo se forman los depósitos de Aβ, cómo se propagan, o cómo poder evitar que adquieran conformaciones alternativas, entre otros. Por último, y más importante, hay que realizar estos estudios en nuevos modelos murinos que representen mejor el avance de la enfermedad de estadio tardío, ya que, permitirá una mejor traslacionalidad de los resultados desde la investigación básica a sus aplicaciones clínicas.
AGRADECIMIENTOS
Este trabajo ha sido financiado por el Ministerio de Ciencia e Innovación (proyecto PID2019-108911RA-100 a DBV), la Asociación de Alzheimer (proyecto AARG-22-9282/9 a DBV), el programa Beatriz Galindo (contrato BAGAL18/00052 a DBV), el Instituto de Salud Carlos III (proyecto PI21/00915 a AG) cofinanciado con fondos FEDER de la Unión Europea y por la Consejería de Economía y Conocimiento de la Junta de Andalucía (proyecto P18-RT-2233 a AG) cofinanciado por el programa operativo FEDER 2014-2020. Las figuras de este artículo han sido elaboradas en biorender.com.
DECLARACIÓN DE TRANSPARENCIA
Los autores/as de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
BIBLIOGRAFÍA
- ↑Golde TE. Alzheimer’s disease: the journey of a healthy brain into organ failure. Mol Neurodegener. 2022; 17: 1-19.
- ↑2022 Alzheimer’s disease facts and figures. Alzheimers Dement. 2022; 18(4): 700-789.
- ↑Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019; 179: 312-339.
- ↑Dhillon S. Aducanumab: First approval. Drugs. 2021; 81(12): 1437-1443.
- ↑McDade E, Cummings JL, Dhadda S et al. Lecanemab in patients with early Alzheimer’s disease: Detailed results on biomarker, cognitive, and clinical effects from the randomized and open-label extension of the phase 2 proof-of-concept study. Alzheimers Res Ther. 2022; 14(1): 1-17.
- ↑Jucker M, Walker LC. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat Neurosci. 2018; 21: 1341-1349.
- ↑Gómez-Gutiérrez R, Morales R. The prion-like phenomenon in Alzheimer’s disease: Evidence of pathology transmission in humans. PLoS Pathog. 2020; 16(10): 1-6.
- ↑Knopman DS, Amieva H, Petersen RC et al. Alzheimer disease. Nat Rev Dis Primers. 2021; 7(1): 1-47.
- ↑Li X, Ospitalieri S, Robberechts T et al. Seeding, maturation and propagation of amyloid β-peptide aggregates in Alzheimer’s disease. Brain. 2022; 145(10): 3558-3570.
- ↑Ulm BS, Borchelt DR, Moore BD. Remodeling Alzheimer-amyloidosis models by seeding. Mol Neurodegener. 2021; 16: 1-11.
- ↑Boon BDC, Bulk M, Jonker AJ et al. The coarse-grained plaque: a divergent Aβ plaque-type in early-onset Alzheimer’s disease. Acta Neuropathol. 2020; 140(6): 811-830.
- ↑Xu G, Ran Y, Fromholt SE et al. Murine Aβ over-production produces diffuse and compact Alzheimer-type amyloid deposits. Acta Neuropathol Commun. 2015; 3: 72.
- ↑Zhou R, Yang G, Guo X et al. Recognition of the amyloid precursor protein by human gamma-secretase. Science. 2019; 363(6428): 1-8.
- ↑Hoogmartens J, Cacace R, van Broeckhoven C. Insight into the genetic etiology of alzheimer’s disease: a comprehensive review of the role of rare variants. Alzheimers Dement. 2021; 13(1): 1-14.
- ↑Lau HHC, Ingelsson M, Watts JC. The existence of Aβ strains and their potential for driving phenotypic heterogeneity in Alzheimer’s disease. Acta Neuropathol. 2021; 14: 17-39.
- ↑Parhizkar S, Arzberger T, Brendel M et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci. 2019; 22(2); 191-204.
- ↑McAllister BB, Lacoursiere SG, Sutherland RJ, Mohajerani MH. Intracerebral seeding of amyloid-β and tau pathology in mice: Factors underlying prion-like spreading and comparisons with α-synuclein. Neurosci and Biobehav Rev. 2020; 11: 1-27.
- ↑Durán-Aniotz C, Moreno-González I, Gámez N et al. Amyloid pathology arrangements in Alzheimer’s disease brains modulate in vivo seeding capability. Acta Neuropathol Commun. 2021; 9(56): 1-13.
- ↑Morales R, Bravo-Alegría J, Moreno-González I et al. Transmission of cerebral amyloid pathology by peripheral administration of misfolded Aβ aggregates. Mol Psychiatry. 2021; 26(10): 5690-5701.
- ↑Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med. 2016; 8(6): 595-608.
- ↑Calderón-Garcidueñas AL, Duyckaerts C. Alzheimer disease. Handb Clin Neurol. 2018; 145: 325-337.
- ↑Busche MA, Hyman BT. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat Neurosci. 2020; 23(10): 1183-1193.
- ↑Gibbons GS, Lee VMY, Trojanowski JQ. Mechanisms of cell-to-cell transmission of pathological tau: a review. JAMA Neurol. 2019; 76(1): 101-108.
- ↑Holtzman DM, Carrillo MC, Hendrix JA et al. Tau: From research to clinical development. Alzheimers Dement. 2016; 12(10): 1033-1039.
- ↑Hardy J, Higgins G. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992; 256(5054): 184-185.
- ↑Moreno-González I, Soto C. Misfolded protein aggregates: Mechanisms, structures and potential for disease transmission. Semin Cell Dev Biol. 2011; 22(5): 482-487.
- ↑Morales R, Durán-Aniotz C, Castilla J et al. De novo induction of amyloid-β deposition in vivo. Mol Psychiatry. 2012; 17(12): 1347-1353.
- ↑Morales R, Durán-Aniotz C, Bravo-Alegría J et al. Infusion of blood from mice displaying cerebral amyloidosis accelerates amyloid pathology in animal models of Alzheimer’s disease. Acta Neuropathol Commun. 2020; 8(213): 1-16.
- ↑Meyer-Luehmann M, Coomaraswamy J, Bolmont T et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006; 313(5794): 1781-1784.
- ↑Eisele YS, Obermüller U, Heilbronner G et al. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science. 2010; 330(6006): 980-982.
- ↑Hamaguchi T, Eisele YS, Varvel NH et al. The presence of Aβ seeds, and not age per se, is critical to the initiation of Aβ deposition in the brain. Acta Neuropathol. 2012; 123(1): 31-37.
- ↑Friesen M, Meyer-Luehmann M. Aβ Seeding as a tool to study cerebral amyloidosis and associated pathology. Front Mol Neurosci. 2019; 12(233): 1-9.
- ↑Condello C, Lemmin T, Stöhr J et al. Structural heterogeneity and intersubject variability of Aβ in familial and sporadic Alzheimer’s disease. Proc Natl Acad Sci USA. 2018; 115(4): E782–E791.
- ↑Makowski L. The structural basis of amyloid strains in Alzheimer’s disease. ACS Biomater Sci Eng. 2020; 6(5): 2498-2505.
- ↑Tian Y, Meng L, Zhang Z. What is strain in neurodegenerative diseases? Cell Mol Life Sci. 2020; 77(4): 665-676.
- ↑Morales R, Moreno-González I, Soto C. Cross-seeding of misfolded proteins: implications for etiology and pathogenesis of protein misfolding diseases. PLoS Pathog. 2013; 9(9): 1-4.
- ↑Stöhr J, Watts JC, Mensinger ZL et al. Purified and synthetic Alzheimer’s amyloid beta (Aβ) prions. Proc Natl Acad Sci USA. 2012; 109(27): 11025-11030.
- ↑Bu XL, Xiang Y, Jin WS et al. Blood-derived amyloid-β protein induces Alzheimer’s disease pathologies. Mol Psychiatry. 2018; 23(9): 1948-1956.
- ↑Ridley RM, Baker HF, Windle CP, Cummings RM. Very long term studies of the seeding of β-amyloidosis in primates. J Neural Transm. 2006; 113(9): 1243-1251.
- ↑Ritchie DL, Adlard P, Peden AH et al. Amyloid-β accumulation in the CNS in human growth hormone recipients in the UK. Acta Neuropathol. 2017; 134(2): 221-240.
- ↑Baglietto-Vargas D, Forner S, Cai L et al. Generation of a humanized Aβ expressing mouse demonstrating aspects of Alzheimer’s disease-like pathology. Nat Commun. 2021; 12(1): 1-16.
- ↑Trujillo-Estrada L, Sánchez-Mejías E, Sánchez-Varo R et al. Animal and cellular models of Alzheimer’s disease: Progress, promise, and future approaches. Neuroscientist. 2022; 28(6): 572-593.
David Baglietto Vargas
Dpto. Biología Celular, Genética y Fisiología. Facultad de Ciencias
Campus de Teatinos. Blvr. de Louis Pasteur, S/N Universidad de Málaga · 29071 Málaga
Tlf.: +34 952 131 647 | E-Mail: d.baglietto@uma.es
viado: 16.11.22
Revisado: 25.11.22
Aceptado: 14.12.22