

Palabras clave: Histiocitosis; Mielofibrosis; BRAF V600E; Vemurafenib.
Keywords: Histyocytosis; Myelofibrosis; BRAF V600E; Vemurafenib.
Introducción
La enfermedad de Erdheim-Chester (ECD) es un tipo poco común de histiocitosis que se incluye en la familia de histiocitosis no relacionadas con las células de Langerhans (1). Desde su descripción inicial en 1930 por los patólogos Erdheim y Chester (2) se han reportado un pequeño número de casos. Sin embargo, en los últimos 10-15 años, gracias a una mejor comprensión de la fisiopatología y las manifestaciones clínicas de la enfermedad el número de casos presentados ha aumentado significativamente.
La ECD es causada por una infiltración histiocitaria que muestra un perfil inmunohistoquímico característico: CD68+, CD163+ y CD1a-. Prácticamente cualquier órgano puede verse afectado por la enfermedad, lo que da lugar a un amplio espectro de expresiones clínicas. A pesar de que la ECD puede permanecer indolente durante años, a veces muestra una progresión rápida y puede ser fatal en cuestión de meses.
Debido a la relativa escasez de casos descritos, la epidemiología de la enfermedad sigue siendo incierta. Los pacientes suelen ser hombres, en la quinta o sexta década de la vida y, curiosamente, se ha encontrado relación con pacientes con neoplasias mieloides, principalmente en síndromes mieloproliferativos crónicos y síndromes mielodisplásicos (3,4,5).
Caso clínico
Presentamos el caso de un varón de 66 años diagnosticado en 1991 de trombocitemia esencial, tratado con hidroxiurea y anagrelida. En 2005, a raíz de nuevas citopenias el paciente fue diagnosticado de una progresión a mielofibrosis CALR+ y en 2012 fue incluido en un ensayo clínico con el inhibidor JAK1/JAK2 ruxolitinib.
En marzo de 2019, el paciente fue ingresado en el hospital con insuficiencia renal aguda y fiebre. Durante la hospitalización, el paciente refirió síntomas B desde hacía aproximadamente un mes. La tomografía computarizada (TC) mostró un aumento de la esplenomegalia (de 15 a 22 cm) con imágenes de infartos esplénicos. Los cultivos de sangre y orina fueron estériles. El ecocardiograma no mostró signos de insuficiencia cardíaca o pericarditis.
Los análisis de laboratorio mostraron un empeoramiento de la bicitopenia (Hb: 82 g/L y plaquetas: 14×10^6/L). El empeoramiento clínico y los hallazgos de laboratorio llevaron a un estudio de médula ósea, que mostró una infiltración masiva por histiocitos espumosos (Figuras 1 y 2). El perfil de inmunohistoquímica mostró positividad para CD58KP1, CD68PG-M1, CD163 y FXIIIa, siendo CD1a y S100 negativos (Figura 3). Además, se detectó la mutación BRAFV600E mediante estudio molecular.
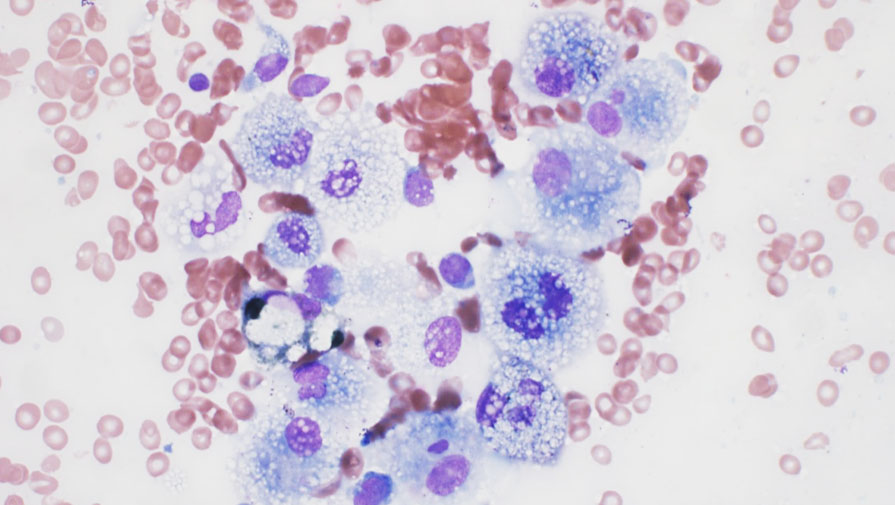
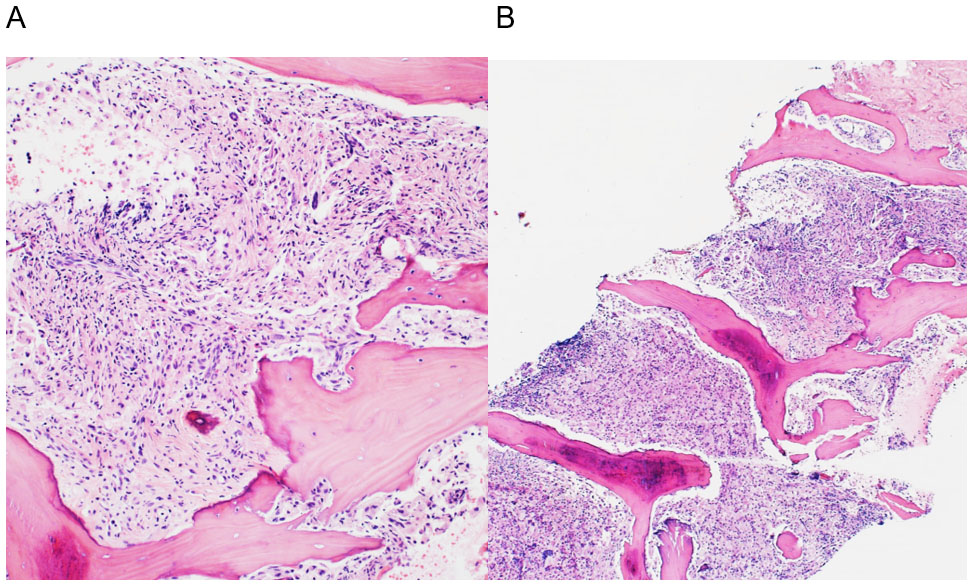
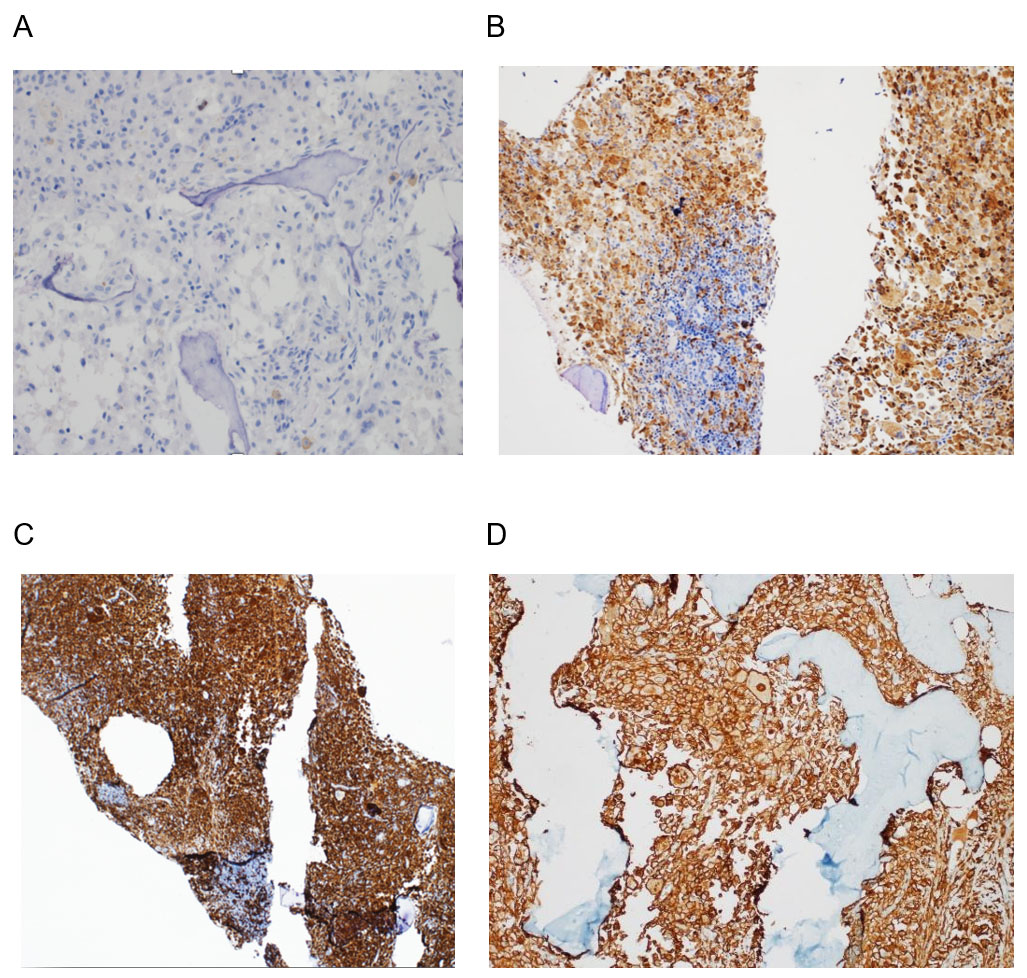
Basándonos en los hallazgos morfológicos, inmunohistoquímicos y moleculares el paciente fue diagnosticado de ECD y se solicitó el tratamiento con el inhibidor de la vía BRAF, vemurafenib. Sin embargo, no se pudo iniciar debido al rápido deterioro del paciente, que resultó en su fallecimiento solo 6 días después del ingreso.
Discusión
El paciente presenta una forma poco común de ECD, ya que en el momento del diagnóstico no estaban presentes ninguna de las manifestaciones clínicas más frecuentes de la enfermedad, que incluyen dolor óseo y osteosclerosis en las pruebas radiológicas, afectación cardiovascular con la imagen clásica de la aorta revestida o derrame pericárdico, afectación renal que produce insuficiencia renal con la imagen de “riñones peludos”, y afectación neurológica, cutánea o pulmonar (6).
Este paciente representa un caso de difícil diagnóstico debido a las múltiples posibilidades de diagnóstico diferencial. Aunque no contamos con una PET-TC, ninguna de las manifestaciones radiológicas más frecuentes estaba presentes (7,8).
A pesar de ello, el paciente pudo ser diagnosticado debido a los hallazgos histopatológicos en la biopsia de médula ósea. El diagnóstico se basó en criterios inmunohistológicos, como la presencia de histiocitos CD68+ asociados con fibrosis y células gigantes Touton, así como los hallazgos moleculares, con la detección de la mutación BRAFV600E (6).
El punto más interesante del caso es la coexistencia de la ECD con una neoplasia mieloide (NM) y la evolución rápida y fatal. Un estudio de Papo et al., en una cohorte de 189 pacientes con ECD, mostró que el 10% de estos pacientes habían sido previamente diagnosticados con alguna forma de NM (4). Se ha demostrado que la ECD está relacionada con frecuencia con la leucemia mielomonocítica crónica (3), pero también se ha asociado con policitemia vera y, como en el caso que presentamos, con la trombocitemia esencial (5,8).
En la mutación BRAFV600E, una valina se sustituye por un ácido glutámico en la posición 600 de la proteína (gen cambio de T>A en el nucleótido 1799). Este cambio representa la mutación más común en la vía MAPK, que representa hasta el 76% de las mutaciones en esta vía (4). BRAF participa en la fosforilación de uno de los puntos más relevantes de la vía. Esta mutación provoca una proliferación celular descontrolada en la línea germinal mieloide (10). La mutación BRAFV600E también puede encontrarse en otros procesos malignos, como el melanoma, el carcinoma papilar de tiroides y en la tricoleucemia.
En la mayoría de los casos revisados en la literatura, los pacientes que desarrollaron una ECD asociada a una NM mostraron otras mutaciones diferentes de BRAFV600E, como ALK, KRAS, NRAS o PIK3C. En esos casos, los pacientes eran de mayor edad y tenían un peor pronóstico (3).
Antes de la aprobación de vemurafenib, el abordaje de tratamiento para la ECD incluía esteroides, vincristina y el trasplante autólogo de células hematopoyéticas (6).
En noviembre de 2017, fue aprobado por la EMA el inhibidor de BRAF llamado vemurafenib, previamente utilizado para el tratamiento del melanoma, para su uso en monoterapia en la ECD. Esta aprobación se basó en los resultados del ensayo VE-BASKET (10), en el cual, a pesar del bajo número de pacientes incluidos, este inhibidor mostró una buena tasa de respuesta global (54.5%).
A pesar de que la enfermedad de Erdheim-Chester (ECD) es una afección poco común con un amplio espectro de manifestaciones clínicas, se debe tener en cuenta, especialmente en pacientes con una evolución inusual de una neoplasia mieloproliferativa. Cuando se sospecha, el diagnóstico histológico y molecular es fundamental. En pacientes sintomáticos, el tratamiento precoz es esencial para tratar de evitar un desenlace fatal, es por ello que hay que sospecharlo y diagnosticarlo lo antes posible.
DECLARACIÓN DE TRANSPARENCIA
Los autores/as de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
BIBIOGRAFÍA
- ↑Khoury, J.D., Solary, E., Abla, O. et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022;36: 1703–1719 .
- ↑W. Chester. Lipoidgranulomatose. Virchows Arch Pathol Anat. 1930; 279:561-602.
- ↑Bonnet P, Chasset F, Moguelet P, et al. Erdheim-Chester disease associated with chronic myelomonocytic leukemia harboring the same clonal mutation. Haematologica 2019; 104(11):530-533.
- ↑Papo M, Diamond EL, Cohen-Aubart F, et al. High prevalence of myeloid neoplasms in adults with non-Langerhans cell histiocytosis. Blood 2017; 130(8):1007-1013.
- ↑Iurlo A, Dagna L, Cattaneo D, et al. Erdheim-Chester Disease with Multiorgan Involvement, Following Polycythemia Vera. Med (United States). Medicine 2016; 95(20):1-4.
- ↑Diamond EL, Dagna L, Hyman DM, et al. Consensus guidelines for the diagnosis and clinical management of Erdheim-Chester disease. Blood 2014; 124(4):483-492.
- ↑Estrada-Veras JI, O’Brien KJ, Boyd LC, et al. The clinical spectrum of Erdheim-Chester disease: An observational cohort study. Blood Adv. 2017; 1(6):357-366.
- ↑Merai H, Collas D, Bhagat A, Mandalia U. Erdheim-Chester Disease : A Case Report and Review of the Literature. Journal of clinical Imaging Science 2020; 10(37):1-5.
- Haroun F, Millado K, Tabbara I. Erdheim-Chester disease: Comprehensive review of molecular profiling and therapeutic advances. Anticancer Res. 2017; 37(6):2777-2783.
- ↑Diamond EL, Subbiah V, Craig Lockhart A, et al. Vemurafenib for BRAF V600-mutant erdheim-chester disease and langerhans cell histiocytosis analysis of data from the histology-independent, phase 2, open-label VE-BASKET study. JAMA Oncol. 2018; 4(3):384-388.
Jose Maria Aspa Cilleruelo
Hospital Universitario Príncipe de Asturias, Alcalá de Henares
Av. Principal de la Universidad, s/n
28805 · Alcalá de Henares, Madrid
Año 2023 · número 140 (03) · páginas 305 a 308
Enviado: 10.11.23
Revisado: 19.11.23
Aceptado: 15.12.23