Resumen
El linfoma folicular (LF) es el segundo linfoma no Hodgkin (LNH) más común y con diferencia el linfoma indolente o de bajo grado de malignidad más frecuente. Es un tumor de células centro germinales con patrón folicular, mixto y rara vez difuso, que habitualmente se presenta en estadíos avanzados y que evoluciona en picos y valles, con supervivencias largas, siendo el tiempo entre pico y pico cada vez más corto, en sucesivas líneas de tratamiento.
Existen varios sistemas de evaluación pronóstica, pero es muy desfavorable la resistencia primaria al tratamiento o la recaída temprana antes de 2 años, que ocurre en el 20% de los enfermos. En la actualidad no existe modo de identificar de manera individual los pacientes que desarrollarán recidiva precoz.
Existe una dificultad para consensuar la secuencia de las distintas líneas de terapia, incluyendo la primera opción de tratamiento. También es destacable la ausencia de estudios comparativos aleatorizados de los tratamientos innovativos. Como primera línea de tratamiento en el linfoma folicular en estadío I y algunos pacientes en el estadío II se utiliza la radioterapia en campo afecto, que puede ser curativa en el 50% de los casos. En estadíos avanzados la variabilidad terapéutica oscila entre la observación o eventualmente monoterapia con Rituximab en pacientes asintomáticos y con bajo volumen de tumor. En los casos sintomáticos o con alto volumen tumoral, la terapia habitual consiste en la administración de quimioinmunoterapia frecuentemente con la combinación de Bendamustina y Rituximab (BR). En los pacientes con linfoma folicular de grado histológico 3b se suele utilizar CHOP-R (Ciclofosfamida, Adriamicina, Vincristina, Prednisona y Rituximab), debido a la similitud de estos casos con el linfoma B difuso de célula grande. La terapia de mantenimiento con Rituximab (R) mejora la supervivencia libre de progresión, pero no es claro que influya sobre la supervivencia global.
Como tratamiento de segunda línea existe una gran diversidad de terapias en la práctica clínica. Cabe citar la quimioterapia o quimioinmunoterapia empleando agentes citotóxicos previamente no utilizados, y la inmunoterapia con la combinación Lenalidomida/Rituximab. En todos los pacientes “jóvenes” y quimiosensibles se puede recurrir al trasplante autólogo de progenitores hematopoyéticos.
Para la tercera línea de tratamiento existe una oferta numerosa. Básicamente el mayor papel terapéutico en la actualidad es atribuible al empleo de anticuerpos biespecíficos, terapia con CAR-T Cells- anti-CD19 o eventualmente a procedimientos de trasplante autólogo o alogénico. Su papel definitivo se conocerá con los resultados de los estudios actualmente en desarrollo. Muchos de los pacientes diagnosticados de linfoma folicular en la época actual tienen una expectativa de vida superior a los 20 años.Abstract
Follicular lymphoma (FL) is the second most common non-Hodgkin lymphoma and by far the most frequent indolent or low-grade malignancy lymphoma. It is a tumor of germinal center cells with a follicular, mixed, and rarely diffuse pattern, which usually presents in advanced stages and evolves in peaks and valleys, with long survivals, although the time between peaks becomes increasingly shorter with successive lines of treatment.
There are several prognostic evaluation systems, but primary resistance to treatment or early relapse before 2 years, which occurs in 20% of patients, is very unfavorable. Currently, there is no way to individually identify patients who will develop early relapse.
There is a difficulty in reaching a consensus on the sequence of different lines of therapy, including the first treatment option. It is also notable the absence of randomized comparative studies of innovative treatments. As a first-line treatment for stage I follicular lymphoma and for some patients in stage II, involved field radiotherapy is used, which can be curative in 50% of cases. In advanced stages, therapeutic variability ranges from observation in asymptomatic patients with low tumor volume to, in symptomatic cases or those with high tumor volume, the administration of Rituximab or alternatively chemoimmunotherapy, often with the combination of Bendamustine and Rituximab (BR). In patients with grade 3b histological follicular lymphoma, CHOP-R (Cyclophosphamide, Adriamycin, Vincristine, Prednisone, and Rituximab) is usually used, due to the similarity of these cases to diffuse large B-cell lymphoma. Maintenance therapy with Rituximab improves progression-free survival, but it is not clear if it affects overall survival.
As a second-line treatment, there is a wide variety of therapies in clinical practice. These include chemotherapy or chemoimmunotherapy using previously unused cytotoxic agents, and immunotherapy with the combination of Lenalidomide/Rituximab. In all “young” and chemosensitive patients, autologous hematopoietic stem cell transplantation can be considered.
For third-line treatment, there are numerous options. Currently, the most significant therapeutic role is attributed to the use of bispecific antibodies, CAR-T Cells-anti-CD19 therapy, or eventually to autologous or allogeneic transplant procedures. In any case, many of the patients diagnosed with follicular lymphoma in the current era have a life expectancy of over 20 years.Palabras clave: Linfoma folicular; Quimioinmunoterapia; Inmunoterapia; Anti-CD20; Tratamiento de mantenimiento; Trasplante autólogo de progenitores Hematopoyéticos; Inmunoterapia T; Anticuerpos biespecíficos.
Keywords: Follicular lymphoma; Chemoimmunotherapy; Immunotherapy; Anti-CD20; Maintenance treatment; Autologous hematopoietic stem cell transplant; T-cell immunotherapy; Bispecific antibodies.
INTRODUCCIÓN
El linfoma folicular (LF) es el segundo linfoma no Hodgkin más común y representa aproximadamente el 25-30% de los linfomas no Hodgkin. Por otro lado, es el linfoma indolente o de bajo grado de malignidad más frecuente. Entendemos por indolencia la ausencia de necesidad terapéutica durante años en este tipo de pacientes. Esta variedad de linfoma es de etiología desconocida y la edad media en el momento del diagnóstico está comprendida generalmente entre los 60 y 70 años. Es igual la incidencia en ambos sexos. Existe el LF en todas las razas, en todas las localizaciones geográficas y muy ocasionalmente se observan casos familiares.
Recientemente, se ha publicado la 5ª edición de la clasificación de los tumores hematolinfoides por parte de la OMS (Organización Mundial de la Salud) con respecto al LF (1). Se distingue el LF clásico que supone el 85% de los casos, con histología nodular, presencia de centrocítos y centroblastos y presenta la t(14;18) en el 85% de los enfermos. Se han distinguido 3 subtipos: el LF con modelo predominantemente folicular, el LF con signos citológicos variables y el LF de célula grande B. Otras variedades bien tipificadas son el LF in situ (2), el pediátrico (3), el duodenal (4) y el primariamente cutáneo (5) (Tabla 1). Esta clasificación representa un aspecto de la variabilidad y heterogeneidad existente dentro del LF. Nos vamos a referir en este artículo al LF clásico.

CARACTERISTICAS CLÍNICO-PATOLÓGICAS
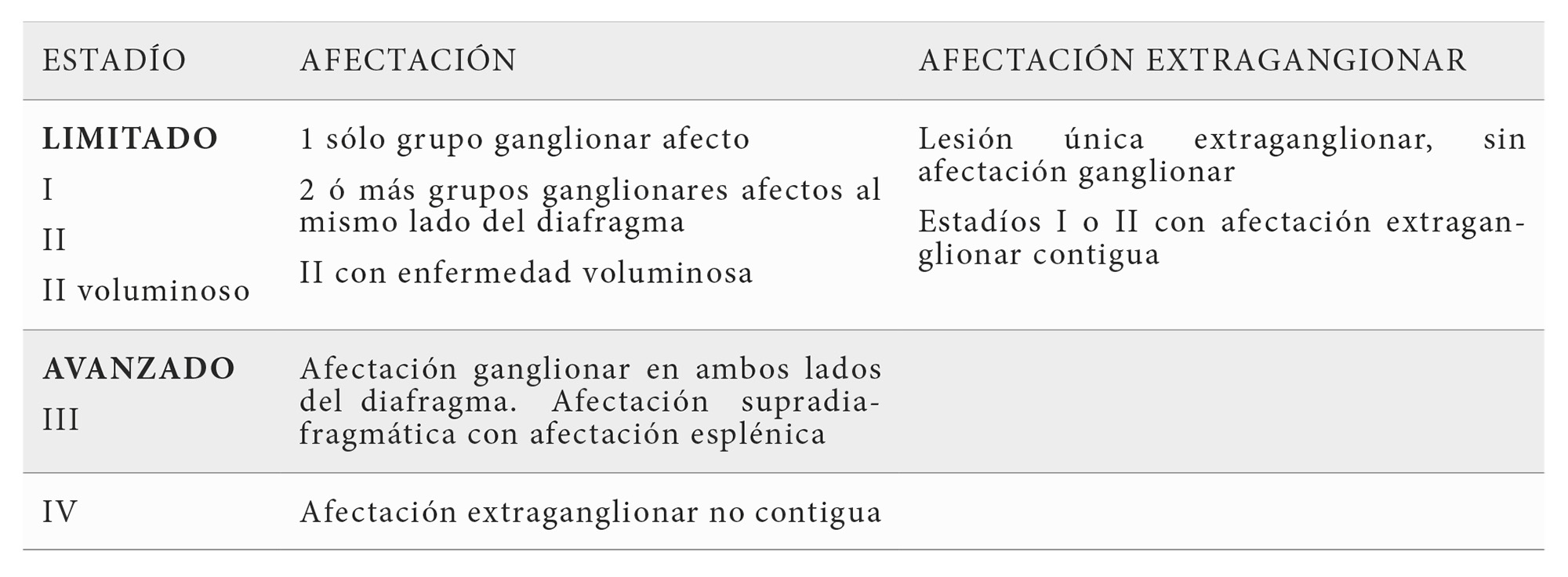
Desde el punto de vista clínico la mayoría de los casos inicialmente son asintomáticos y debutan con linfadenopatía exclusivamente. Solo una pequeña proporción de pacientes muestran en el momento del diagnóstico síntomas sistémicos, tal como la fiebre, pérdida de peso o sudoración nocturna. El LF se localiza fundamentalmente en el ámbito ganglionar y es extremadamente rara la afectación primaria extraganglionar. La mayor parte de los pacientes presentan desde el momento del diagnóstico enfermedad diseminada, debutando por tanto en estadios clínicos avanzados (Tabla 2). Los enfermos muestran habitualmente linfoadenopatía supra e infradiafragmática, siendo rara la afectación mediastínica. La enfermedad en ocasiones se presenta con masas ganglionares abdominales, que pueden ser un hallazgo en exploraciones practicadas por otro motivo. Es frecuente la hepatomegalia, la esplenomegalia y la infiltración de la médula ósea con patrón paratrabecular y que puede ocurrir hasta 70-80% de los enfermos (6).
¿Cómo realizar el diagnóstico?
El diagnóstico se efectúa por biopsia ganglionar, a ser posible con extirpación de un ganglio afecto. En ausencia de ganglios superficiales se puede recurrir a la biopsia con aguja gruesa guiada radiológicamente y cuando ello no es posible se debe de practicar por cirugía.
Si los cortes histológicos son adecuados, un hemopatólogo podría realizar el diagnóstico solamente con la morfología en la mayoría de los casos. Sin embargo, la práctica de la inmunohistoquímica es útil en algunos pacientes y la combinación de ambos procedimientos se ha convertido en un estándar para el diagnóstico del LF. Este linfoma es un tumor de células centrogerminales, es decir, de centrocitos o linfocitos pequeños hendidos y de centroblastos o linfocitos grandes no hendidos. De manera característica el patrón es típicamente nodular, en algunas ocasiones mixto y muy rara vez difuso. Desde el punto de vista histológico se distinguen 3 grados de acuerdo con la proporción de centroblastos. El grado 1 se caracteriza por la práctica ausencia de células grandes que suponen menos del 5% en el campo microscópico. En el grado 2 el número de centroblastos está comprendido entre 5-15%. La presencia de centroblastos es muy relevante en el grado 3, clasificándose este en dos subtipos: el 3a, donde existe presencia de centrocitos y el 3b compuesto por una infiltración masiva de centroblastos. Recientemente, el valor clínico de la distinción en grados histológicos ha suscitado algunas controversias. Los casos grado 1, 2 y 3a tienen el mismo comportamiento clínico, portan las mismas alteraciones genéticas y muestran similar respuesta terapéutica. El grado 3b se asimila tanto clínica como histológicamente al linfoma B difuso de célula grande (7). No es fácil la diferenciación entre ambas entidades y también distinguirlo de una eventual evolución de un LF clásico, a un linfoma no Hodgkin B transformado de célula grande. Independientemente, el valor clínico de los grados histológicos se ha reducido ostensiblemente, básicamente por la deficiente reproductibilidad entre distintos patólogos.
La inmunohistoquímica, tiene un papel muy importante para una correcta catalogación de los linfomas y para el diagnóstico diferencial. Los datos inmunohistoquímicos en el LF, se caracterizan fundamentalmente por la existencia de marcadores B del centrogerminal y ausencia de marcadores T. Las células del LF habitualmente muestran inmunoglobulinas de superficie, en la mayoría de los casos IgM, en menor cuantía IgG y excepcionalmente IgA. Las cadenas Kappa y Lambda se detectan, pero nunca las dos. Las células del LF expresan HLA-DR y antígenos B como CD19, CD20, CD79a, CD21 y CD10. Habitualmente no muestran CD23. Son siempre CD5 negativos salvo casos excepcionales. La tinción citoplasmática de Bcl2 en el LF clásico es positiva, en contraste a otros linfomas foliculares como el cutáneo, el infantil y el grado 3b.
El típico marcador cromosómico del LF clásico es la t(14;18) que aparece en el 85% de los pacientes (8). Se trata de una traslocación entre el brazo largo del cromosoma 18, lugar del oncogen Bcl2 y uno de los 3 lugares del gen de las inmunoglobulinas. Frecuentemente la traslocación ocurre con el gen de las cadenas pesadas situado en el cromosoma 14, resultando entonces la t(14;18). En algunas ocasiones la traslocación ocurre con el gen de la cadena Kappa situado en el cromosoma 2 resultando entonces la t(2;18) y a veces con el gen de las cadenas Lambda, situado en el cromosoma 22 resultando la t(18;22). La consecuencia de estas traslocaciones es un alto nivel de la proteína bcl2, que produce una resistencia a la apoptosis, es decir, a la muerte celular programada. La t(14,18) no es específica del LF y puede observarse en otros linfomas, en casos de hiperplasia folicular reactiva y en algún ganglio de sujetos aparentemente normales que nunca o muy excepcionalmente desarrollan linfoma. Algunos casos de LF muestran anomalías en el cromosoma 3 afectando al bcl6. El reordenamiento bcl6 es más frecuente en pacientes con LF de grado histológico 3b.
Algunos matices en la evaluación del enfermo diagnosticado de LF
Como es lógico, la historia clínica, la exploración física, los datos hematológicos, bioquímicos, inmunológicos, serológicos, y la beta-2 microglobulina, forman parte estándar de la evaluación del paciente.
¿Es necesaria la práctica sistemática de la biopsia de la médula ósea, como se ha venido haciendo hasta épocas recientes? La médula ósea está involucrada en la enfermedad en el 70-80% de los casos, pero su valor clínico es muy limitado (9,10). Por un lado no contribuye al pronóstico, ni tampoco a la respuesta al tratamiento en el 99% de los casos y también por el motivo de la mayor sensibilidad de la inmunoterapia con monoclonales anti-CD20 para eliminar la enfermedad en la médula ósea. Por ello, la práctica rutinaria de la biopsia de médula ósea es innecesaria y solo debe de realizarse en casos sospechosos de padecer LF estadio clínico I definido por PET-TAC. En esta situación sí es relevante el resultado de la biopsia medular, ya que los estadios clínicos I “auténticos” por PET-TAC y biopsia de médula ósea son potencialmente curables en un 50-70% de casos mediante la radioterapia en campo afecto.
Otra indicación de la práctica de biopsia de médula ósea es el estudio de las citopenias, que bien pueden deberse a infiltración o alternativamente a hiperesplenismo o ser de patogenia autoinmune.
¿Se debe practicar TAC o PET-TAC para el estadiaje o el control evolutivo del paciente con LF? (11,12) Si clínicamente el paciente puede ser candidato a encontrarse en estadio clínico I, el PET-TAC es imprescindible como método conjuntamente con la biopsia de médula ósea para tratar de asegurar con los mejores medios, que el paciente se encuentra en dicho estadío. Si el enfermo se presenta como es habitual en el 70-80% de los casos en estadíos avanzados, se debe de practicar TAC, no solo en la evaluación sino en los futuros controles evolutivos. Si existe sospecha de transformación, que ocurre a un ritmo de 2% anual y se detecta en una alta proporción de casos en estudios de necropsia, es relevante la práctica del PET-TAC, porque al reflejar un cambio claro en la actividad metabólica celular del tumor, contribuye con otros datos tal como síntomas sistémicos, crecimiento rápido ganglionar, afectación extraganglionar, incremento de la LDH, a la sospecha clínica de transformación a célula grande que debe de verificarse mediante nueva biopsia ganglionar.
El diagnóstico diferencial debe de realizarse fundamentalmente con la hiperplasia folicular reactiva, el linfoma folicular cutáneo, el linfoma B de células grades rico en células T, el linfoma del manto y el linfoma de zona marginal. Las diferencias pueden establecerse en base a datos clínicos, morfológicos, inmunohistoquímicos, y genéticos en estos procesos linfoproliferativos.
VALORACIÓN PRONÓSTICA
El LF es típicamente un linfoma de bajo grado de malignidad, que generalmente se presenta con linfadenopatía asintomática, a veces fluctuante y que sigue un curso indolente sin necesidad de tratamiento durante años en muchos pacientes. Aunque el curso evolutivo de LF es variable, con mucha frecuencia los enfermos sobreviven más de 20 años. Esto no siempre es así y existe una minoría de casos cercano al 20% con evolución claramente desfavorable, bien con ausencia de respuesta al tratamiento primario o con recaída temprana antes de los 2 años.
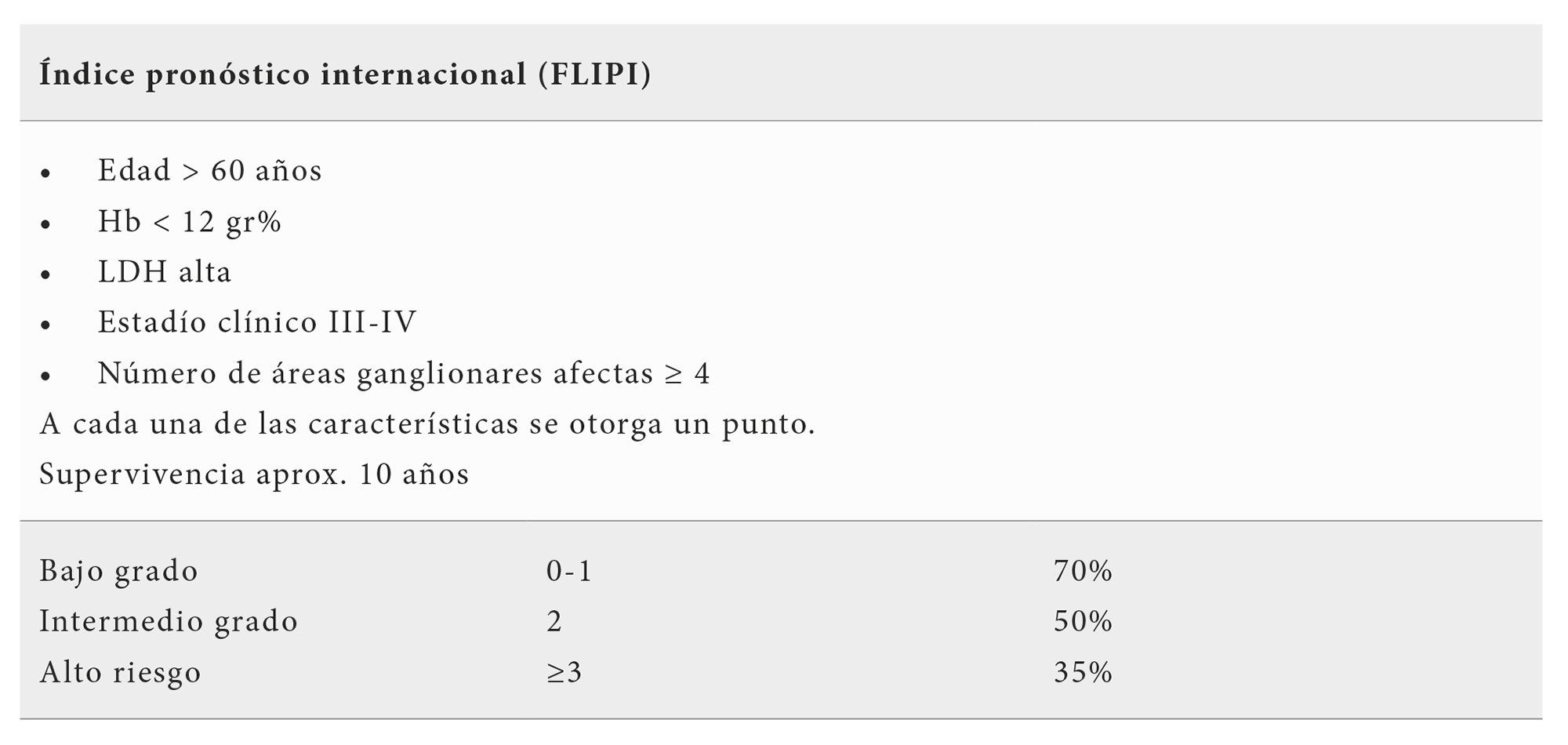
El sistema de evolución pronóstica más generalizado es el denominado FLIPI (Tabla 3) (índice pronóstico internacional del linfoma folicular) cuyo valor ha sido probado en la época preinmunoterápica y también en pacientes que han recibido tratamiento con Rituximab solo o en combinación (13). Se han identificado 5 factores adversos que son: edad > 60 años, estadío clínico III o IV, nivel de Hb < 12 gr/dl, 4 o más áreas ganglionares afectas y tasa de LDH elevada. Otorgando un punto a cada una de estas características se han distinguido 3 grupos de riesgo: alto riesgo con 3 o más factores adversos, grupo intermedio con 2 factores adversos y bajo riesgo, 0 ó 1 factores adversos. Existen diferencias entre estos grupos en la supervivencia de libre enfermedad y en la supervivencia global.
Otro índice pronóstico como el PRIMA utiliza la beta-2 microglobulina y la infiltración de médula ósea para definir 3 grupos. En principio, este sistema parece ser menos discriminativo (14). No existe un método para identificar al paciente con resistencia primaria o recaída precoz antes de los 2 años de aplicar la primera línea de tratamiento. Tras la 1ª recaída del LF el factor pronóstico más importante es la duración de la primera remisión.
¿CUÁNDO TRATAR?
Como en muchos procesos de curso mayoritariamente indolente el momento de iniciar el tratamiento es una decisión relevante. Está perfectamente demostrado que una terapia temprana en pacientes asintomáticos con linfadenopatía poco voluminosa, no prolonga la vida, aunque si puede suponer un incremento en la supervivencia libre de enfermedad y un retraso en la administración de la quimioinmunoterapia. ¿Es valorable esto en un paciente asintomático? Algunos pacientes asintomáticos y con enfermedad ganglionar diseminada y poco voluminosa pueden mostrar algún grado de incertidumbre, cierta ansiedad y desean ser tratados rápidamente. En estos casos se debe realizar una valoración clínica individual y si se acuerda con el paciente un tratamiento, ofrecer una terapia monovalente con Rituximab.
En síntesis, la observación es la actitud más comúnmente seguida en los enfermos con LF asintomáticos y con linfadenopatía poco voluminosa.
El tratamiento debe de iniciarse cuando exista progresión ganglionar marcada o enfermedad voluminosa, compromiso de la función de un órgano, aparición de síntomas sistémicos, evidencia de afectación extraganglionar, presencia de citopenias bien por infiltración, autoinmunidad o hiperesplenismo (Tabla 4).

TRATAMIENTO: Oferta, controversias y perspectivas
Tratamiento inicial o de primera línea
Como hemos señalado previamente no hay una postura uniforme acerca del tratamiento inicial del LF. El tratamiento se debe de seleccionar en función de los estadíos clínicos y del grado histológico. Comúnmente se aplica un tratamiento similar a los enfermos con grado histológico 1, 2 y 3a y reciben una atención terapéutica distinta los pacientes cuyo grado histológico es 3b.
Los estadíos I evaluados mediante PET-TAC y biopsia de médula ósea deben de ser tratados con radioterapia en campo afecto. Ordinariamente se aplican 24 Gy y la cifra de curaciones obtenidas oscilan entre el 50 y el 70% de los enfermos (15).
Algunos grupos tratan el estadío II de manera similar a los estadíos más avanzados III y IV. Sin embargo, otros equipos aplican en casos seleccionados dentro del estadío II la misma opción terapéutica que en el estadío I, es decir, la radioterapia en campo afecto. Una minoría de enfermos padece el LF estadío clínico II en regiones donde es factible la radioterapia en campo afecto con toxicidad limitada y pueden ser candidatos a la misma. La probabilidad de curación con radioterapia en el estadío II del LF es real, pero claramente inferior a la obtenida en el tratamiento del estadío I. La adición de un curso corto de quimioterapia o inmunoquimioterapia ha logrado aumentar la supervivencia libre de enfermedad, pero no ha afectado a la supervivencia global.
En los pacientes con enfermedad diseminada y asintomáticos en el momento del diagnóstico, que constituyen la mayoría de los casos, puede optarse por la observación en los que presentan linfadenopatía poco voluminosa. Como previamente hemos comentado algunos enfermos asintomáticos y con linfadenopatía diseminada, pero poco voluminosa, demandan tratamiento de manera rápida. En estas circunstancias se puede administrar Rituximab en terapia monovalente. La pauta más generalmente empleada consiste en la administración semanal de Rituximab durante 4 semanas, seguida o no de una terapia de mantenimiento con Rituximab bimensual durante 2 años. Tras una primera aplicación del Rituximab por vía intravenosa se puede continuar con Rituximab subcutáneo (Rituximab/Hyaluronidasa) de eficacia similar y administración más cómoda para el paciente (16). En pacientes con LF y hepatitis C concomitante es conveniente tratar el virus C, antes de acometer el tratamiento del linfoma. Este fármaco puede propiciar la reactivación de la hepatitis B en enfermos positivos para el antígeno de superficie o anticuerpos frente al antígeno “core” de la hepatitis B.
Un evento tóxico excepcionalmente raro es la leucoencefalopatía multifocal progresiva en relación con el virus JC.
En aquellos pacientes en los que existe una clara indicación terapéutica puede optarse por aplicar solo Rituximab, o bien por una combinación de quimio/inmunoterapia, usualmente con la asociación Bendamustina/Rituximab (BR) o de manera alternativa con la combinación Bendamustina/Obinutuzumab (BO) (17,18). Esta última pauta aunque equivalente en términos de eficiencia terapéutica es menos empleada por su mayor toxicidad. En el estudio GALLIUM se evidenció que la combinación BO producía una mayor supervivencia libre de progresión, aunque una similar supervivencia global y probabilidad de transformación a célula grande que la pauta BR.
Otra posibilidad en casos sintomáticos, es el empleo de la combinación Lenalidomida/Rituximab (19,20) aunque su papel en nuestro ámbito está más limitado a una segunda línea de tratamiento. Esta combinación es capaz de producir una supervivencia libre de progresión semejante a la observada con un régimen de quimioinmunoterapia, siendo diferente la toxicidad. En el ensayo de fase 3 RELEVANCE se comparó la combinación de Lenalidomida/Rituximab a la quimioinmunoterapia, pudiéndose aplicar quimioterapias distintas CHOP-R, BR o CVP-R, según la experiencia de cada centro. Se observaron cifras equiparables de remisiones completas, siendo similar la supervivencia global, la probabilidad de transformación histológica a los 6 años y también un número semejante de neoplasias secundarias (21).
La terapia inicial en estadíos avanzados, II, III y IV de LF y grado histológico 3b es distinta dado su semejanza clínica e histológica con el linfoma B difuso de célula grande. El tratamiento empleado en estos casos, consiste en quimioterapia combinada e inmunoterapia con un anti-CD20, generalmente Rituximab. Las pautas más utilizadas han sido CHOP-R, CVP-R (Ciclofosfamida, Vincristina, Prednisona y Rituximab). Otros esquemas que contienen Fludarabina han sido abandonados por su mayor toxicidad. La combinación más frecuentemente empleada ha sido el CHOP-R. El CVP-R puede ser una alternativa aceptable cuando existe algún tipo de cardiopatía, aunque otra posibilidad consiste en emplear la Adriamicina liposomal en lugar de la estándar.
Este enfoque terapéutico inicial para el LF ampliamente difundido se expone en la tabla 5.

En la era preinmunoterápica, nuestro grupo utilizó la combinación de quimioterapia tipo CVP asociado a interferón alfa2b en la inducción y el tratamiento de mantenimiento de los linfomas no Hodgkin de bajo grado de malignidad, la mayor parte de ellos con el diagnóstico de linfoma folicular. La adición de interferón alfa-2b a la quimioterapia no mejoró la supervivencia global (22). La introducción en la práctica clínica del Rituximab solo en combinación con quimioterapia supuso un claro avance en la supervivencia de los pacientes.
Terapia de mantenimiento
El 20% de los pacientes con LF presentan una recaída precoz, antes de 2 años y constituyen un serio problema terapéutico. Es muy evidente en todos los pacientes con LF el valor pronóstico de la duración de la primera remisión completa. Se asocian a una remisión corta varios factores: sexo masculino, deficiente estado general, beta-2 microglobulina alta y FLIPI de alto riesgo. No disponemos de biomarcadores que de manera individual identifiquen a estos pacientes. En contraste, en la actualidad y a diferencia con otras hemopatías malignas es discutible el valor en la práctica clínica de la enfermedad mínima residual, medida por PCR para bcl2 en los enfermos tratados por LF (23).
Una vez conseguida la remisión completa mediante un tratamiento de inducción, se ha intentado prolongar al máximo la duración de la misma, administrando una terapia de mantenimiento. Se ha investigado el papel del mantenimiento con Rituximab en el estudio PRIMA (24). El Rituximab se administra cada 2 meses durante 2 años tras el tratamiento de inducción.
Se evidenció que el Rituximab de mantenimiento era capaz de incrementar la supervivencia libre de progresión con respecto a la observación, pero no se pudieron documentar diferencias con respecto a la supervivencia global (25). Existen datos sugestivos acerca del mismo efecto clínico utilizando un mantenimiento con Rituximab más corto, de 4 dosis aplicando una cada 2 meses.
Tratamiento de segunda línea
En la mayoría de los casos tras un periodo de 10 años o más sobreviene la primera recaída. No disponemos de un acuerdo amplio acerca de la segunda línea de tratamiento y existe una evidente variabilidad entre distintas ofertas terapéuticas. Clásicamente en la era preinmunoterápica el tratamiento consistía en quimioterapia combinada incluyendo platino, en nuestra experiencia con el empleo de ESHAP (Etopósido, Metilprednisolona, Cisplatino y Arabinósido de citosina) seguido en caso de quimiosensibilidad de colecta celular y trasplante autólogo de progenitores hematopoyéticos. Este proceder resulta en un alto número de remisiones y larga supervivencia libre de enfermedad en una proporción muy significativa de pacientes, cercano al 50%. Años después con la llegada de la inmunoterapia se añadió el Rituximab al esquema quimioterápico.
Hoy en día una posibilidad como terapia de segunda línea consiste en la aplicación de la pauta Lenalidomida/Rituximab (25). Este esquema está exento de quimioterapia. El ensayo AUGMENT comparó la rama Lenalidomida/Rituximab a otra con Rituximab/Placebo. Se demostró un incremento claro en la supervivencia libre de enfermedad de la primera opción, variando la toxicidad en ambas ramas dada la inclusión de la Lenalidomida en una de ellas.
Se ha estudiado también en un ensayo fase I/II el papel terapéutico de la combinación Lenalidomida/Obinutuzumab (26) en los linfomas indolentes en recaída. Se obtuvo un 72% de remisiones completas y una supervivencia libre de progresión de un 73% a los dos años.
El Tazemetostat (27), es un inhibidor de EZH2, un regulador epigenético mutado en aproximadamente una cuarta parte de los pacientes. Ha sido investigado en un fase II no encontrándose diferencias en la supervivencia libre de progresión entre los casos mutados y no mutados. Es un fármaco aceptablemente tolerado, de eficacia limitada en terapia monovalente y constituye una eventual oferta como segunda línea o posteriores.
Otros fármacos como el Venetoclax y el Ibrutinib han ofrecido resultados en general pobres en el LF (28,29,30,31). Así mismo, los inhibidores de la fosfatidilinositol 3 kinasa (PI3K), como el idelalisib han sido prácticamente abandonados en la práctica clínica por su eficiencia limitada y toxicidad acusada, tal como infección, neumonitis, colitis, hepatotoxicidad y exantema (32).
En síntesis, la combinación Lenalidomida/Rituximab o eventualmente Lenalidomida/Obinutuzumab constituye una opción válida como tratamiento de segunda línea en el LF. Para pacientes “jóvenes” y quimiosensibles, el empleo de quimioinmunoterapia tipo ESHAP-R seguida de trasplante autólogo de progenitores hematopoyéticos constituye una alternativa razonable. No se dispone de estudios comparativos entre ambas opciones terapéuticas.
Tratamiento de tercera línea o posteriores
La oferta terapéutica en esta situación clínica consiste fundamentalmente en la administración de anticuerpos biespecificos, terapia con antiCD9 CAR-T Cells y procedimientos de trasplante autólogo o alogénico.
El anticuerpo biespecífico CD3/CD20 Mosunetuzumab fue aprobado recientemente para el tratamiento en tercera línea del LF. En un estudio multicéntrico que incluyó 90 pacientes se observó la respuesta completa en el 60% de los enfermos. Los efectos secundarios más notables fueron el síndrome de liberación de citoquinas, la neutropenia, la hipofosfatemia, la hiperglucemia, y la anemia. La mediana de la supervivencia libre de progresión a los 3 años ha sido algo superior al 40%. Las perspectivas terapéuticas del Mosunetuzumab mejoran claramente las obtenidas con otros fármacos (33,34,35,36). Actualmente, este anticuerpo biespecífico está siendo estudiado en combinación con Lenalidomida, Polatuzumab y otros fármacos.
Así mismo, otros biespecíficos como Odronextamab y Epcoritamab están siendo investigados en la actualidad (37,38). Ambos son biespecíficos CD3/CD20. El primero de ellos ha sido recientemente aprobado por la EMA para el tratamiento del LF resistente o recidivante y también en las mismas condiciones para el linfoma B difuso de célula grande. En el momento presente el Epcoritamab se encuentra también aprobado por la EMA y FDA para el linfoma B difuso de célula grande refractario o recidivante, tras dos líneas de tratamiento.
La terapia CAR-T Cells anti-CD19 desde hace años desempeña un papel terapéutico importante en el tratamiento del linfoma B difuso de célula grande recidivante. Los estudios en el linfoma folicular son más recientes, pero han conducido a su aprobación como terapia tras dos o más líneas de tratamiento en el LF, con la excepción de Yescarta (Axicell) por la EMA tras 3 líneas de tratamiento. El ensayo ZUMA 5 (39,40), con Axicabtagene, Ciloleucel (Axicel), incluyó una mayoría de pacientes con LF y también enfermos con linfoma de zona marginal. El 94% de los pacientes con LF obtuvieron una respuesta, que fue completa en un 79% y la mediana de supervivencia libre de progresión fue de 40,2 meses. La mediana de supervivencia no se había alcanzado. La toxicidad fue relevante básicamente por el síndrome de liberación de citoquinas, neurotoxicidad, citopenias e infecciones. El ensayo ELARA (41) con Tisagenlecleucel (Tisacel) ofreció resultados similares. Este estudio incluyó pacientes de mayor riesgo y ofreció un 86,2% de respuestas globales incluyendo 69,1% de respuestas completas. El perfil de toxicidad fue algo más favorable que en el ZUMA 5. A falta de una comparación directa, quizá las diferencias estriben en la población de enfermos que participaron en ambos estudios.
En síntesis, puede haber cierto grado de competencia entre los anticuerpos biespecíficos empleados en tratamiento monovalente o quizá en combinación con la terapia CAR-T Cells anti-CD19. No existe una comparación directa entre biespecíficos y CAR-T Cells en pacientes con LF. Por ello, aun no es clara la secuencia de ambos tratamientos, aunque a mi parecer los datos actuales apoyan la “pole position” de la terapia con CAR-T Cells. Se ha mostrado la respuesta a biespecíficos tras la recidiva a tratamiento con CAR-T Cells anti-CD19. No existen datos en el sentido opuesto. En ambos casos, existe abundante información que demuestra su eficacia en el LF en pacientes que han recibido inmunoterapia, quimioinmunoterapia y trasplante autólogo de progenitores hematopoyéticos.
En la Tabla 6 se expone una secuencia de posibles terapias una vez finalizada la inducción.

No existen datos comparativos de la inmunoterapia T frente a los procedimientos de trasplante tanto autólogo como alogénico. El papel del autólogo parece claro en el tratamiento de segunda línea del LF (42). Es posible el carácter curativo del trasplante alogénico en una población de pacientes con LF. La edad y las complicaciones clínicas del alotrasplante han frenado su uso que en la actualidad es minoritario (43). El papel definitivo tanto de los anticuerpos biespecíficos como de la terapia con anti-CD19 CAR-T Cells requiere del conocimiento de muchos de los estudios hoy en desarrollo. El concepto de “curación funcional” hoy en día puede aplicarse a cierta proporción de enfermos con LF. Quizá la “curación real” sea un objetivo próximo con los tratamientos innovativos.
DECLARACIÓN DE TRANSPARENCIA
El autor/a de este artículo declara no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
BIBLIOGRAFÍA
- ↑Kurtz KS, Kalmbach S, Ott M, et al. Follicular Lymphoma in the 5th Edition of the WHO-classification of Haematolymphoid Neoplasms. Updated classification and New Biological Data. Cancers. 2023; 15:785-804.
- ↑Henopp T, Quintanilla-Martínez L, Fend F, et al. Prevalence of follicular lymphoma in situ in consecutively analysed reactive lymph nodes. Histopathology. 2011; 59:139-142.
- ↑Louissaint A, Ackerman AM, Dias-Santagata D, et al. Pediatric-type nodal follicular lymphoma: an indolent clonal proliferation in children and adults with high proliferation index and no BCL2 rearrangement. Blood. 2012; 120:2395-2404.
- ↑Takata K, Okada H, Ohmiya N, et al. Primary gastrointestinal follicular lymphoma involving the duodenal second portion is a distinct entity: a multicenter, retrospective analysis in Japan. Cancer Sci. 2011; 102:1532-1536.
- ↑Zinzani PL, Quaglino P, Pimpinelli N, et al. Prognostic factors in primary cutaneous B-cell lymphoma: The Italian Study Group for Cutaneous Lymphomas. J. Clin. Oncol. 2006; 24:1376-1382.
- ↑Horming SJ, Rosenberg SA. The natural history of initially untreated low-grade non-Hodgkin’s lymphomas. N Eng J Med. 1984; 311:1471-1475.
- ↑Horn H, Schmelter C, Leich E, et al. Follicular lymphoma grade 3B is a distinct neoplasm according to cytogenetic and immunohistochemical profiles. Haematologica. 2011; 96:1327-1334.
- ↑Rowley JD. Chromosome studies in the non-Hodgkin’s lymphomas: the role of the 14;18 translocation. J Clin Oncol. 1988; 6:919-925.
- ↑Rutherford SC, Herold M, Hiddemann W, et al. Impact of bone marrow biopsy on response assessment in immunochemotherapy-treated lymphoma patients in GALLIUM and GOYA. Blood Adv. 2020; 4:1589-1593.
- ↑Rutherford SC, Li V, Ghione P, et al. Bone marrow biopsies do not impact response assessment for follicular lymphoma patients treated on clinical trials. Br J Haematol. 2017; 179:242-245.
- ↑Cheson BD, Fisher RI, Barrington SF, et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014; 32:3059-3068.
- ↑Barrington SF, Mikhaeel NG, Kostakoglu L, et al. Role of imaging in the staging and response assessment of lymphoma: consensus of the International Conference on Malignant Lymphomas Imaging Working Group. J Clin Oncol. 2014; 32:3048-3058.
- ↑Solal-Céligny P, Roy P, Colombat P, et al. Follicular lymphoma international prognostic index. Blood. 2004; 104:1258-1265.
- ↑Kimby E, Lockmer S, Holte H, et al. The simplified follicular lymphoma PRIMA Prognostic index is useful in patients wiht first-line chemo-free Rituximab-based therapy. Br J Haematol. 2020; 191:738-747.
- ↑Friedberg JW, Byrtek M, Link BK, et al. Effectiveness of first-line management strategies for stage I follicular lymphoma: analysis of the National LymphoCare Study. J Clin Oncol. 2012; 30:3368-3375.
- ↑Cartron G, Bachy E, Tilly H, et al. Randomized Phase III Trial Evaluating Subcutaneous Rituximab for the First-Line Treatment of Low-Tumor Burden Follicular Lymphoma: Results of a LYSA Study. J Clin Oncol. 2023; 41:3523-3533.
- ↑Townsend W, Hiddemann W, Buske C, et al. Obinutuzumab versus Rituximab Immunochemotherapy in Previously Untreated iNHL: Final Results From the GALLIUM Study. Hemasphere. 2023; 30;7(7):e919.doi.
- ↑Hiddemann W, Barbui AM, Canales MA, et al. Immunochemotherapy With Obinutuzumab or Rituximab for Previously Untreated Follicular Lymphoma in the GALLIUM Study: Influence of Chemotherapy on Efficacy and Safety. J Clin Oncol. 2018; 36:2395-2404.
- ↑Strati P, Jain P, Johnson RJ, et al. Long-term follow-up of lenalidomide and rituximab as initial treatment of follicular lymphoma. Blood. 2021; 137:1124-1129.
- ↑Bachy E, Houot R, Feugier P, et al. Obinutuzumab plus lenalidomide in advanced, previously untreated follicular lymphoma in need of systemic therapy: a LYSA study. Blood. 2022; 139:2338-2346.
- ↑Morschhauser F, Nastoupil L, Feugier P, et al. Six-Year Results From RELEVANCE Lenalidomide Plus Rituximab (R2) Versus Rituximab-Chemotherapy Followed by Rituximab Maintenance in Untreated Advanced Follicular Lymphoma. J Clin Oncol. 2022; 40:3239-3245.
- ↑Arranz R, García-Alfonso P, Sobrino P, et al. Role of interferon alfa-2b in the induction and maintenance treatment of low-grade non-Hodgkin’s lymphoma: results from a prospective, multicenter trial with double randomization. Journal of Clinical Oncology. 1998; 16:1538-1546.
- ↑Luminari S, Manni M, Galimberti S, et al. Response-Adapted Postinduction Strategy in Patients With Advanced-Stage Follicular Lymphoma: The FOLL12 Study. J Clin Oncol. 2022; 40:729-739.
- ↑Salles G, Seymour JF, Offner F, et al. Rituximab maintenance for 2 years in patients with high tumour burden follicular lymphoma responding to rituximab plus chemotherapy (PRIMA): a phase 3, randomised controlled trial. Lancet. 2011; 377:42-51.
- ↑Leonard JP, Trneny M, Offner F, et al. AUGMENT: A Phase III Study of Lenalidomide Plus Rituximab Versus Placebo Plus Rituximab in Relapsed or Refractory Indolent Lymphoma. J Clin Oncol. 2019; 37:1188-1199.
- ↑Fowler NH, Nastoupil LJ, Chin C. et al. A Phase I/II Study of Lenalidomide Plus Obinutuzumab in Relapsed Indolent Lymphoma. Blood 2019; 134 (Supplement_1): 348.
- ↑Morschhauser F, Tilly H, Chaidos A, et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: An open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020; 21:1433-1442.
- ↑Zinzani PL, Flinn LW, Yuen SLS, et al. Venetoclax-rituximab with or without bendamustine vs bendamustine-rituximab in relapsed/refractory follicular lymphoma. Blood. 2020; 136:2628-2637.
- ↑Barlett NL, Costello BA, LaPlant BR, et al. Single-agent ibrutinib in relapsed or refractory follicular lymphoma: A phase 2 consortium trial. Blood. 2018; 131:182-190.
- ↑Fowler NH, Nastoupil L, De Vos S, et al. The combination of ibrutinib and rituximab demonstrates activity in first-line follicular lymphoma. Br J Haematol. 2020; 189:650-660.
- ↑Otham T, Rosenberg AS, Timmerman J, et al. A Phase II Trial of the Combination of Obinutuzumab, Venetoclax, and Ibrutinib in Patients with Previously Untreated Follicular Lymphoma. Blood 2022; 140 (Supplement 1): 11954–11955.
- ↑Richardson NC, Kasamon Y, Pazdur R, et al. The saga of PI3K inhibitors in haematological malignancies: survival is the ultimate safety endpoint. Lancet Oncol. 2022; 23:563-566.
- ↑Lopedote P, Shadman M. Targeted Treatment of Relapsed or Refractory Follicular Lymphoma: Focus on the Therapeutic Potential of Mosunetuzumab. Cancer Manag Res. 2023; 15:257-264.
- ↑Budde LE, Sehn LH, Matasar M, et al. Safety and efficacy of mosunetuzumab, a bispecific antibody, in patients with relapsed or refractory follicular lymphoma: a single-arm, multicentre, phase 2 study. Lancet Oncol. 2022; 23:1055-1065.
- ↑Barlett NL, Sehn LH, Matasar MJ, et al. Mosunetuzumab Monotherapy Demonstrates Durable Efficacy with a Manageable Safety Profile in Patients with Relapsed/Refractory Follicular Lymphoma Who Received ≥2 Prior Therapies: Updated Results from a Pivotal Phase II Study. Blood 2022; 140 (Supplement 1): 1467–1470.
- ↑Bosch F, Kuruvilla J, Vassilakopoulos TP, et al. Indirect Treatment Comparisons of Mosunetuzumab With Third-and Later-Line Treatments for Relapsed/Refractory Follicular Lymphoma. Clin Lymphoma Myeloma Leuk. 2024;24:105-121.
- ↑Kim TM, Taszner M, Cho SG, et al. Odronextamab in Patients with Relapsed/Refractory (R/R) Follicular Lymphoma (FL) Grade 1-3a: Results from a Prespecified Analysis of the Pivotal Phase II Study ELM-2. Blood 2022; 140 (Supplement 1): 2280–2282.
- ↑Hutchings M, Mous R, Clausen MR, et al. Dose escalation of subcutaneous epcoritamab in patients with relapsed or refractory B-cell non-Hodgkin lymphoma: an open-label phase 1/2 study. Lancet. 2021; 398:1157-1169.
- ↑Jacobson CA, Chavez JC, Sehgal AR, et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol. 2022; 23:91-103.
- ↑Neelapu SS, Chavez JC, Sehgal AR, et al. Three-year follow-up analysis of axicabtagene ciloleucel in relapsed/refractory indolent non-Hodgkin lymphoma (ZUMA-5). Blood. 2024; 143:496-506.
- ↑Fowler NH, Dickinson M, Dreyling M, et al. Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: the phase 2 ELARA trial. Nat Med. 2022; 28:325-332.
- ↑Jiménez-Ubieto A, Grande C, Caballero D et al. Autologous Stem Cell Transplantation for Follicular Lymphoma: Favorable Long-Term Survival Irrespective of Pretransplantation Rituximab Exposure. Biol Blood Marrow Transplant. 2017; 23:1631-1640.
- ↑Sureda A, Zhang MJ, Dreger P, et al. Allogeneic hematopoietic stem cell transplantation for relapsed follicular lymphoma: A combined analysis on behalf of the Lymphoma Working Party of the EBMT and the Lymphoma Committee of the CIBMTR. Cancer. 2018; 124:1733-1742.
ranm tv
José María Fernández-Rañada de la Gándara
Real Academia Nacional de Medicina de España
C/ Arrieta, 12 · 28013 Madrid
Tlf.: +34 91 159 47 34 | E-Mail: jmranada@yahoo.es
An RANM. 2025;142(01): 41-50
Enviado*: 04.02.25
Revisado: 10.02.25
Aceptado: 24.02.25
* Fecha de lectura en la RANM