La exploración y el descubrimiento de nuevos enlaces químicos es de gran importancia para la química, los materiales, la catálisis y campos relacionados. Las interacciones no covalentes han desempeñado un papel importante en muchas áreas de vanguardia de la química moderna, como el diseño de materiales y la biología molecular. En este última caso, es crucial la investigación sobre el origen físico y el alcance de interacciones como los enlaces por puente de hidrógeno, o las interacciones de apilamiento pi···pi.
En un trabajo reciente, Hirao et al. (Chem. Soc. Rev. 2020, 49, 7602-7626), propusieron y analizaron la expansión del alfabeto genético y sus posibles aplicaciones. Ahora, nuestro objetivo es comprender la alta selectividad que muestran algunos de los pares de bases no naturales o artificiales que no presentan interacción de enlaces por puente de hidrógeno con el par de bases complementario, a diferencia de los pares de bases Watson-Crick. Queremos comprender si la incorporación en la cadena del cebador está determinada por el ajuste geométrico; o está determinada únicamente por las interacciones de apilamiento tipo pi. Un trabajo anterior del grupo (Chem. Comm. 2011, 47, 7326-7328) mostró la fuerza impulsora de los enlaces de hidrógeno, pero este no es el caso cuando no están presentes. Concretamente, examinamos la afinidad de varias bases y complejos plantilla-cebador con 1,3-difluorotolueno (F, un isóster de timina), que fue encontrada experimentalmente para incorporarse correctamente en complejos plantilla-cebador, formando pares A-F, en presencia de la ADN polimerasa. Nuestros cálculos mostraron que la afinidad por F es claramente mucho menor que por T. Demostramos que la incorporación correcta de isósteros no polares no se puede explicar sin invocar un mecanismo adicional, como el ajuste estérico del nuevo par de bases en el bolsillo del sitio activo de la polimerasa. La comprensión de estas interacciones debería permitir sentar las bases para el diseño futuro de nuevos pares de bases artificiales.
Por lo tanto, mediante el uso de la química cuántica, nuestro objetivo ha sido comprender la naturaleza electrónica del reconocimiento molecular en pares de bases de ADN artificiales para poder diseñar racionalmente otros pares de bases con el potencial de sufrir autoorganización de forma controlada y selectiva. El estudio se ha centrado en la comprensión de las interacciones orbitalarias donante-aceptor y en las interacciones electrostáticas.
En este trabajo, se seleccionó un grupo de pares de bases de ADN artificiales (Figura 1). Todas ellas fueron propuestas anteriormente y probadas experimentalmente en estudios de replicación.
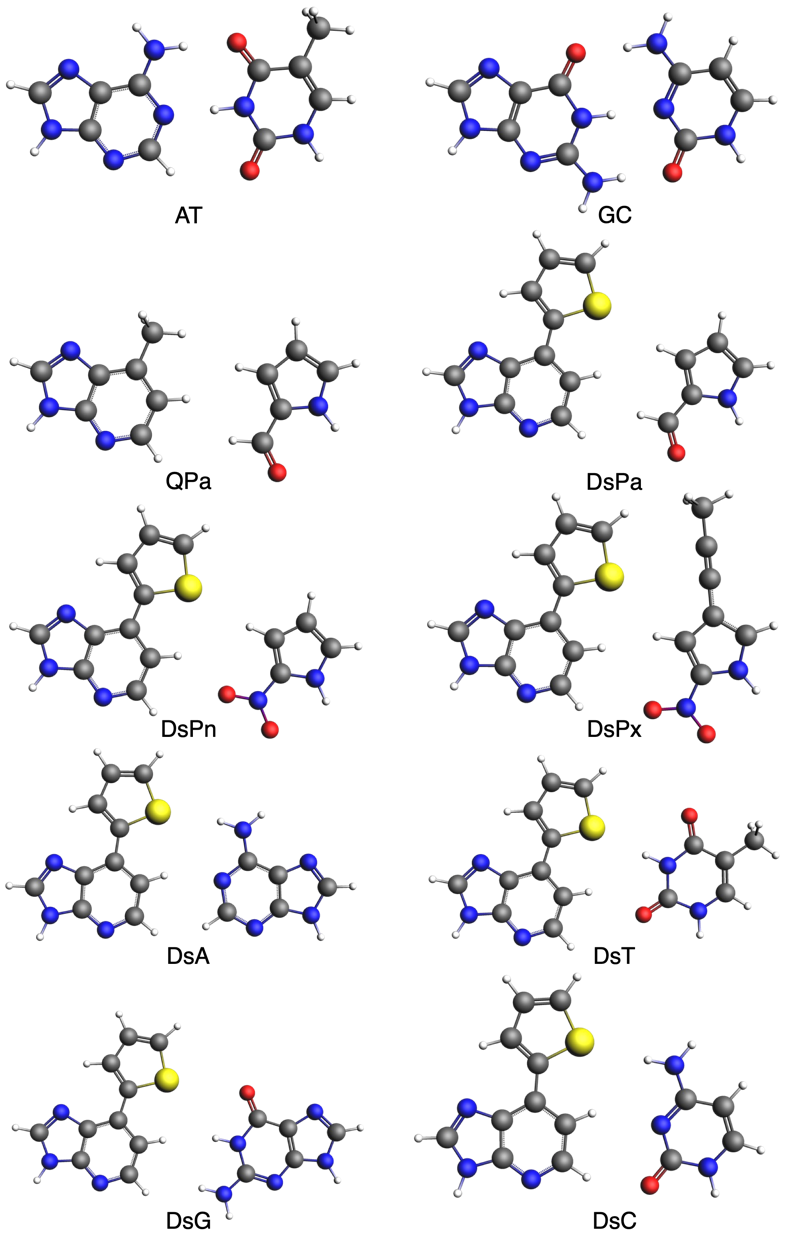
El objetivo es entender su selectividad, y compararla con aquella de los pares de bases Watson-Crick. Hemos demostrado que, a pesar de no presentar interacción por enlaces por puente de hidrógeno, su selectividad viene determinada por las interacciones de apilamiento pi-pi con el par de bases contiguo. Nuestro modelo computacional permite obtener con gran similitud el orden de selectividad observado en el laboratorio. Con los cual, justifica su uso para futuros trabajos con nuevas bases propuestas a la espera de ser sintetizadas para aplicaciones concretas.
Agradecimientos
Soporte económico del Ministerio de Ciencia, Innovación y Universidades (PID2022-138861-I00 y María de Maeztu CEX-2021-001202-M), y la Generalitat de Catalunya (2021SGR442).