Resumen
La Hb Lansing es una hemoglobinopatía hereditaria causada por la mutación en la posición 87 de la cadena alfa de la hemoglobina (Hb), afectando la unión del átomo de hierro. Provoca niveles bajos de SatO2 en pulsioximetría, con PaO2 normal en gasometría arterial, y podría comprometer la estabilidad de la hemoglobina. Se presenta el caso de una mujer chilena de 25 años con cianosis desde la infancia y antecedentes familiares similares, cuya mutación fue confirmada en heterocigosis. Existen pocas publicaciones sobre esta variante, siendo la mayoría de los casos asintomáticos. Este estudio destaca la importancia de considerar la Hb Lansing y otras variantes de Hb con alteraciones de SatO2, no solo como interferencias en la medición con pulsioximetría, sino también como posibles causas de inestabilidad estructural de la hemoglobina y cianosis periférica.
Abstract
Hb Lansing is a hereditary hemoglobinopathy caused by a mutation at position 87 of the alpha chain of hemoglobin (Hb), affecting the binding of the iron atom. It leads to low SatO2 levels on pulse oximetry, with normal PaO2 in arterial blood gas analysis, and may compromise hemoglobin stability. This study presents the case of a 25-year-old Chilean woman with childhood-onset cyanosis and a family history of similar symptoms, whose mutation was confirmed in heterozygosity. There are few publications on this variant, with most reported cases being asymptomatic. This study highlights the importance of considering Hb Lansing and other Hb variants with altered SatO2, not only as interferences in pulse oximetry measurements but also as potential causes of hemoglobin structural instability and peripheral cyanosis.
Palabras clave: Hb Lansing; Hemoglobinopatía de cadena alfa; Pulsioximetría; Cianosis.
Keywords: Hb Lansing; Hemoglobinopathy of alpha globin; Pulse oximetry; Cyanosis.
INTRODUCCIÓN
Las hemoglobinopatías estructurales o variantes de hemoglobina (Hb) son un grupo de trastornos hereditarios autosómicos que se caracterizan por alteraciones cualitativas en las cadenas de alfa o beta globina.
En ellas, existe un cambio de aminoácido o aminoácidos que condicionan cambios en la solubilidad, estabilidad o afinidad por el oxígeno de la Hb, siendo los responsables de las manifestaciones clínicas de estas entidades. La Hb Lansing es una hemoglobinopatía estructural caracterizada por un cambio de histidina por glutamina en el codón 87 del 2º exón del gen HBA2 de globina [α87(F8) His>Gln; HBA2:c.264C>G)], que condiciona niveles bajos de saturación de oxígeno (SatO2) venosa, medida mediante pulsioximetría, con niveles normales en gasometría arterial (PaO2). El primer caso fue descrito en 2009 por Sarikoda (1), donde demostró la presencia de dicha mutación en heterocigosis en un padre y su hija con niveles bajos de SatO2 venosa con PaO2 y P50 normales. A continuación, presentamos un caso de Hb Lansing estudiado en nuestro centro y realizaremos una revisión bibliográfica de casos ya publicados.
Caso Clínico
Se trata de una mujer chilena de 25 años en el momento del estudio, con antecedentes de cianosis detectada a los 4 años con SatO2 del 85% en pulsioximetría de manera permanente. Se realiza un amplio estudio que descarta patología cardiológica y neumológica, siendo manejada con oxigenoterapia hasta los 20 años sin mejoría clínica y con persistencia de SatO2 bajas. Entre sus antecedentes personales destacan idéntica sintomatología en su padre y único hermano, que presentan SatO2 basales de 84-85%.
En marzo de 2024 es remitida a Hematología de su centro de origen por gestación en curso de 15 semanas, para valoración. Se realiza estudio analítico que refleja Hb 11,1 g/dL, hematocrito 33,4%, volumen corpuscular medio 81,9 fL, metahemoglobina 0,4% (valor de referencia 0-1,5%). No se pudo realizar P50. Se realiza estudio de electroforesis capilar de Hb, donde se observa una Hb variante (9% del total) que migra, en pH alcalino, hacia la región de Hb F y Hb S-D; y en pH ácido entre zona S y A-D-E. En ese momento, se ponen en contacto con el Hospital Clínico San Carlos de Madrid, para realizar estudio de hemoglobinopatías. Por cromatografía líquida de alta resolución (HPLC Variant II), se observa un pico en un tiempo de retención (RT) de 1,93 minutos que corresponde al 9,3% del total de la hemoglobina (Figura 1), mientras que la Electroforesis Capilar (EC) muestra un perfil normal (Hb A 97,4%, HbA2 2,6%). El estudio genético, realizado mediante secuenciación masiva (NGS) con el kit Devyser Thalassemia NGS, identificó la mutación CAC>CAG en el codon 87 del gen HBA2 [α87(F8) His>Gln; HBA2:c.264], la cual fue confirmada mediante secuenciación de Sanger del gen HBA2 (Figura 2).
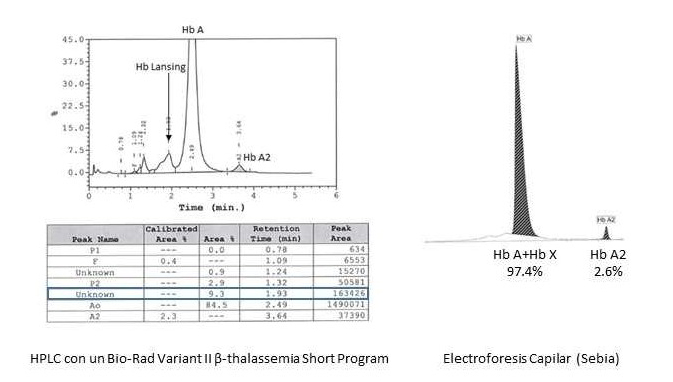
Electroforesis capilar (Sebia) que muestra una única fracción principal que incluye tanto Hb A como la variante (Hb X), representando el 97.4% del total, y una fracción correspondiente a Hb A2 del 2.6%. La co-migración de Hb Lansing con Hb A impide su diferenciación con esta técnica.
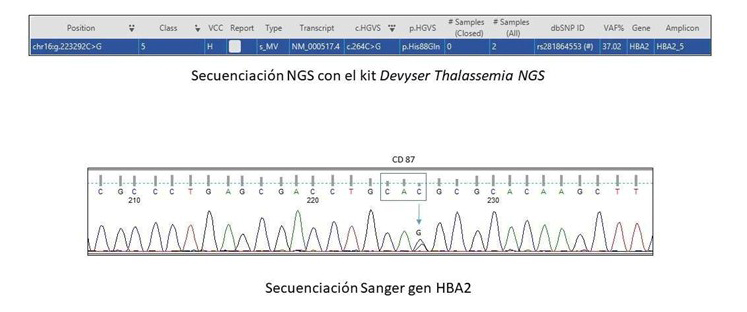
Confirmación de dicha variante mediante secuenciación Sanger del gen HBA2, donde se observa el cambio de base (C>G) en heterocigosidad en el CD87
Discusión
La molécula de hemoglobina consta de 4 subunidades, cada una de las cuales posee un grupo hemo con un átomo de hierro en forma ferrosa, a través del cual cada subunidad es capaz de fijar una molécula de oxígeno. Dentro de la estructura de la hemoglobina las uniones α1β2, los extremos C-terminales de las cadenas β y α, las uniones con el 2,3-DPG en la cavidad central y la cavidad de contacto con el grupo hemo son zonas importantes en la captación del oxígeno y en los efectos alostéricos del 2,3-DPG, CO2 y H+ sobre dicha captación. Por tanto, mutaciones en los aminoácidos comprendidos en estas zonas críticas para la función de la hemoglobina pueden determinar alteraciones en la afinidad por el oxígeno. El residuo de histidina en la posición 87 de la cadena alfa es crítico para la unión del átomo de hierro. Por ello, en la Hb Lansing, al sustituir esa histidina por glutamina, se produce una alteración de la oxihemoglobina y cambio en su absorción a la longitud de onda. Esto explicaría las discrepancias entre la SatO2 venosa medida mediante pulsioximetría y la PaO2 en gasometría arterial. Sin embargo, nuestra paciente presenta además cianosis, por lo que esta mutación también podría implicar repercusiones a nivel funcional o en la estabilidad de la molécula de la hemoglobina.
Hemos realizado una revisión de la literatura y únicamente hemos encontrado 7 publicaciones a nivel mundial sobre esta hemoglobinopatía (1,2,3,4,5), de las cuales 2 son de origen español (6, 7). Existen otras variantes de Hb producidas por mutaciones en el mismo locus que la Hb Lansing con diferentes resultados clínicos. Tanto la Hb Iwata (His→Arg) (8) como la Hb Grifton (His→Pro) no tienen efectos clínicos. Sin embargo, la Hb M-Iwate (His→Tyr) presenta una baja afinidad por el oxígeno con aumento de los niveles de metahemoglobina y cianosis (9).
No existen muchos datos reportados acerca de la P50 de los diferentes casos. En los que sí se refleja, presentan valores dentro del rango de la normalidad, en torno a 25-27 mmHg (1). La mayoría de estos pacientes son asintomáticos y son diagnosticados en edad adulta, comenzando el estudio por hallazgo casual de SatO2 venosa baja en pulsioximetría. Por el contrario, existen pocos casos de pacientes, sobre todo en edad adulta, que presenten cianosis periférica. Los pulsioxímetros emiten luz a longitudes de onda conocidas (660 y 990 nm) y miden la absorbancia de ésta, asumiendo que se realiza por las moléculas de oxihemoglobina y desoxihemoglobina en diferente proporción. Por ello, las Hb variantes pueden interferir en las mediciones del pulsioxímetro debido al diferente espectro de absorción de luz a esas longitudes de onda.
Nuestro caso pone en evidencia la importancia de considerar la Hb Lansing y otros casos de Hb variantes en el bolsillo proximal de unión del átomo de hierro al grupo hemo como, no solamente causa de falsos niveles de SatO2 venosa, sino como posibilidad de inestabilidad estructural de la hemoglobina y cianosis periférica.
No hemos podido realizar en nuestro caso la P50 debido al envío de la muestra desde Santiago de Chile, pero es muy probable que la P50 esté incrementada con descenso de la afinidad de la Hb por el oxígeno.
No puede explicarse con precisión porque unos casos con Hb Lansing presentan cianosis y otros no, es posible que otros factores no bien conocidos intervengan en la producción de cianosis como el estrés oxidativo u otros.
DECLARACIÓN DE TRANSPARENCIA
Los autores/as de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
BIBLIOGRAFÍA
- ↑Sarikonda KV, Ribeiro RS, Herrick JL, Hoyer JD. Hemoglobin Lansing: a novel hemoglobin variant causing falsely decreased oxygen saturation by pulse oximetry. Am J Hematol 2009;84:541. https://doi.org/10.1002/ajh.21452
- ↑Ishitsuka K, Uchino J, Kato J, et al. First reported case of hemoglobin Lansing in Asia detected by false low oxygen saturation on pulse oximetry. Int J Hematol 2012;95:731–732. https://doi.org/10.1007/s12185-012-1096-8.
- ↑Hassan SM, Harteveld CL, Bakker E, Giordano PC. Hb Lansing (HBA2: c.264C > G) and a new β promoter transversion [-52 (G > T)]: an attempt to define the phenotype of two mutations found in the Omani population. Hemoglobin 2015;39:111–114. https://doi.org/10.3109/03630269.2015.1016615.
- ↑Trakulsrichai S, Panthan B, Jittorntam P, et al. First identification of hemoglobin Lansing-Ramathibodi [α87(F8)His → Gln; CAC>CAG (HBA1: C.264C>G)] in a thai family with spurious hypoxemia. Southeast Asian J Trop Med Public Health 2016;47(5):1048–1054.
- ↑Akar N, Torun D, Oztürk A. Hemoglobin Lansing (alpha) [HBA2 CD87 (HIS>GLU) (C>A)] in a Turkish individual resulting from another nucleotide substitution. Turk J Hematol 2014;31:317–318. https://doi.org/10.4274/tjh.2014.0102.
- ↑Fernández Barge T, Sánchez Escamilla M, Domínguez García JJ, Fernández González de Villambrosía I, Muruzabal Siges MJ, López Duarte M. PO-082. Descripción de cuatro casos de hemoglobinopatía Lansing en Cantabria. Sangre 2024;43(Supl 1):176. ISSN: 0036-4355.
- ↑Sarrat-Pirla A, Quilis-Esquerre J, Morales-Sànchez M, Velàzquez-Cerdà M, Uriz-Urzainqui S, Villalba T. Hemoglobina Lansing. Una variant de l’hemoglobina causant de baixa saturació d’oxigen detectada per pulsioximetria. Pediatr Catalana. 2018;78(3):111-113
- ↑Ohba Y, Miyaji T, Hattori Y, et al. Unstable hemoglobins in Japan. Hemoglobin 1980;4:307–312.
- ↑Mayne EE, Elder GE, Lappin TR, Ferguson LA. Hb M Iwate [alpha(2)87His—Tyr beta 2]: De novo mutation in an Irish family. Hemoglobin 1986;10:205–208
Javier Cucharero Martín
Servicio de Hematología. Hospital Clínico San Carlos
C/Profesor Martín Lagos s/n · 28040 Madrid (Spain)
Tlf.: +34 607 294 227 | E-Mail: cucharero.j@gmail.com
An RANM. 2025;142(01): 96-99
Enviado: 24.03.25
Revisado: 30.03.25
Aceptado: 05.04.25