

Resumen
Objetivos. Comprobar con datos hematológicos que se puede establecer el diagnóstico y grado clínico de la β-Talasemia intermedia cuando coexisten una triplicación de genes alfa (αααanti 3.7) y una β-talasemia heterocigota.
Métodos. Estudio retrospectivo en el que participaron 73 pacientes de origen caucásico, que mostraron simultáneamente triplicación o cuadruplicación de genes α y β-talasemia heterocigota.
El cribado de las mutaciones más frecuentes de α-talasemia, así como la triplicación génica (αααanti 3.7) se llevó a cabo mediante PCR multiplex seguida de hibridación inversa y se confirmó mediante MLPA. El diagnóstico molecular de β-talasemia se realizó mediante secuenciación automática según el método de Sanger.
Resultados. Se han clasificado los genotipos en tres grupos según el número de genes α globina y la severidad de la alteración en el gen β globina. Todos presentaron una mutación en el gen β-globina (β0-talasemia, β+-talasemia severa y β+-talasemia leve). El grupo I pacientes que han coheredado 6 genes α y los grupos II y III 5 genes α globina. En el grupo III los pacientes eran portadores de mutaciones que afectan a los genes β y δ globina. Los parámetros hematológicos más significativos fueron los niveles de hemoglobina, el volumen corpuscular medio, el ancho de distribución y el porcentaje de Hb fetal.
Conclusiones. En el grupo I la coheradibilidad de 6 genes alfa globina, ya sea triplicación homocigota (ααα/ααα) o cuadruplicación heterocigota (αααα/αα), con una β-talasemia heterocigótica da como resultado una anemia de severa a moderada que puede requerir de terapia transfusional, siendo la severidad de la mutación del gen β-globina la que determinaría la variación clínica. Los pacientes del grupo II fenotípicamente se comportaron como talasemia intermedia leve. Finalmente, los pacientes del grupo III se comportaron como rasgo talasémico ya que todos eran portadores de mutaciones que aumentan la sobreexpresión de los genes γ.
Abstract
Objectives. Check with hematological data that the diagnosis and clinical grade of β-thalassemia intermedia can be established when a triplication of genes alpha (αααanti 3.7) and heterozygous β-thalassemia are coherent.
Methods. Retrospective study in which 73 patients of Caucasian origin participated, who simultaneously showed a tripling or quadrupling of the genes α and heterozygous β-thalassemia.
Screening for the most frequent α-thalassemia mutations, as well as gene triplication (αααanti 3.7) was carried out by multiplex PCR followed by reverse hybridization and confirmed by MLPA. The molecular diagnosis of β-thalassemia was carried out by automatic sequencing according to the Sanger’s method.
Results. Genotypes have been classified into three groups according to the number of α-globin genes and the severity of the alteration in the β-globin gene. All had a mutation in the β-globin gene (β0-thalassemia, severe β+-thalassemia, and mild β+-thalassemia). Group I patients who have inherited 6 α globin genes. Group II and group III have inherited 5 α globin genes. In group III, the patients were carriers of mutations affecting the β and δ globin genes. The most significant hematological parameters were hemoglobin levels, mean corpuscular volume, red deep width, and percentage of fetal hemoglobin.
Conclusions. In group I, patients who have inherited of 6 α globin genes, either by homozygous triplication (ααα/ααα) or heterozygous quadruplication (αααα/αα), with heterozygous β-thalassemia results in severe to moderate anemia that may require transfusion therapy, being the severity of the β-globin gene mutation that would determine the clinical variation. Group II patients behaved phenotypically like mild thalassemia intermedia. Finally, group III patients behaved like a thalassemic trait since all were carriers of mutations that increase the overexpression of g genes.
Palabras clave: β-talasemia intermedia; Triplicación de genes α; Mutaciones en el gen β globina.
Keywords: β-thalassemia intermedia; α-globin gene triplication; β globin gene mutations.
introducción
Los síndromes talasémicos constituyen las enfermedades genéticas más comunes en todo el mundo. Comprenden un grupo complejo muy heterogéneo de trastornos de la hemoglobina (Hb), caracterizados por el defecto de síntesis de una o más cadenas de globina. Según la cadena de globina que se encuentra disminuida o ausente, se clasifican en α, β, δ, βδ, γδβ o εγδβ. Se heredan de forma autosómica recesiva y se caracterizan por una extrema diversidad de fenotipos, lo que hace que el diagnóstico sea un verdadero reto (1, 2).
El espectro de las β-talasemias es amplio, va desde la β-talasemia mayor (β-TM) que se caracteriza por una anemia severa a partir de los primeros años de vida, erigiéndose como una enfermedad grave transfusión-dependiente, a formas leves, generalmente heterocigotos con anemia microcítica e hipocroma sin manifestaciones clínicas evidentes, conocidas como β-talasemia minor, y formas intermedias [β-talasemia intermedia (β-TI)] no transfusión-dependientes (3, 4).
El término β-TI fue sugerido por vez primera en 1955 para describir pacientes que tenían manifestaciones clínicas que resultaban ser demasiado graves para denominarse β-talasemia minor y a la vez, demasiado leves para llamarse β-TM (5).
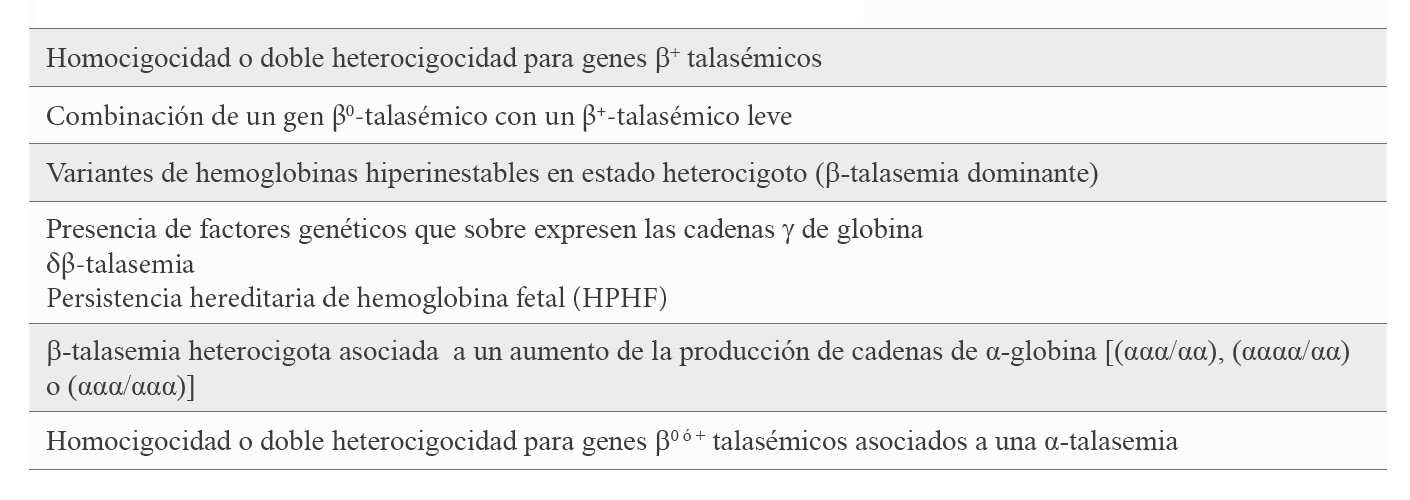
Aunque la β-TI carece de correlación molecular específica y el diagnóstico sigue siendo en gran parte clínico, se ha descrito una asociación genotipo/fenotipo de modo que la base genética de la diversidad fenotípica vendría determinada por moduladores genéticos. La mayoría de los pacientes con β-TI son homocigotos o heterocigotos compuestos para la β-talasemia, lo que significa que ambos loci de β-globina están afectados y la enfermedad tiene un patrón genético recesivo. La amplia diversidad de mutaciones que afecta al gen β-globina (HBB), van desde las mutaciones leves del promotor (β+-talasemia leve) que causan una ligera reducción en la producción de la cadena β-globina, pasando por las β+-talasemias, donde la producción de cadena β-globina está disminuida, hasta las β0-talasemias con ausencia total de cadena β-globina. La heterocigocidad compuesta para estas mutaciones proporciona un amplio espectro de fenotipos clínicos (6). Con menor frecuencia, se observa la afectación de un solo un locus de β-globina, siendo el otro completamente normal; así, en estos casos, la β-TI es de herencia autosómica dominante como en el caso de las hemoglobinas hiperinestables (7). Otros moduladores genéticos son aquellos que están directamente involucrados en el desequilibrio de las cadenas β-globina. La homocigocidad o doble heterocigocidad con una sobre-expresión de las cadenas γ de globina, bien porque uno de los alelos o los dos se correspondan con una δβ-talasemia, o porque se asocian a alteraciones moleculares en el mismo locus o en otro diferente que condicionan un incremento de la síntesis de cadenas γ-globina (8). También el aumento de la producción de cadenas de α-globina por una triplicación o cuadruplicación de genes α (ααα/αα o αααα/αα) asociado con una β-talasemia heterocigota se ha visto que podría cursar como una -TI (9) (Tabla 1).
El objetivo de este trabajo es comprobar, con datos hematimétricos y de manera empírica, si se puede establecer el diagnóstico y grado clínico de la β-TI cuando coexisten una triplicación de genes alfa (αααanti 3.7) y una β-talasemia heterocigota, ya que la triplicación de genes alfa globina es considerado un factor importante en la severidad de la β-talasemia, exacerbando la expresión fenotípica de la misma, al causar un mayor desequilibrio entre las cadenas de globina. Para ello presentamos nuestra experiencia de casos de β-talasemia asociada a una triplicación de genes alfa (αααanti 3.7) en España, en los últimos 10 años en el Hospital Clínico San Carlos de Madrid.
MATERIAL Y MÉTODOS
Este trabajo es un estudio retrospectivo llevado a cabo desde enero de 2010 a diciembre de 2019 y en el que participaron 73 pacientes de origen caucásico, que mostraron simultáneamente triplicación o cuadruplicación de genes α y β-talasemia heterocigota.
Los parámetros hematológicos y el recuento de reticulocitos fueron determinados con un contador automático de células (Coulter LH750 Analyzer; Beckman Coulter, Brea, CA, USA), incluyendo el análisis morfológico celular.
Los niveles de Hb A2 y Hb F fueron medidos por cromatografía líquida de alta resolución (HPLC) (VARIANTTM; Bio-Rad Laboratories, Hercules, CA, USA). Las hemoglobinas fueron analizadas por electroforesis capilar zonal siguiendo las instrucciones del fabricante para el sistema Sebia Capillarys Flex (Sebia, Norcross, GA), y mediante HPLC de intercambio iónico siguiendo las pautas del fabricante para el programa corto de β-talasemia de BioRad Variant II (Bio-Rad, Hercules, CA).
Tras el aislamiento del ADN genómico con un método automático (BiorobotÒ EZ1; Quiagen GmbH, Hilden, Germany), el ADN genómico fue cuantificado con un NanoDrop 1000 (Thermo Scientific, Wilmington, DE, EE. UU).
El cribado de las mutaciones más frecuentes de α-talasemia así como la triplicación génica (αααanti 3.7) se llevó a cabo mediante PCR multiplex seguida de hibridación inversa con un kit comercial Alpha-Globin StripAssay (ViennaLab Diagnostic GmbH, Vienna, Austria) y se confirmó mediante Multiplex Ligation-dependent Probe Amplification (MLPA), este análisis se llevó a cabo utilizando el kit MLPA (SALSA MLPA KIT P140 HBA; MRC Holland, Amsterdam, The Netherlands). El diagnóstico molecular de β-talasemia se realizó mediante secuenciación automática según el método de Sanger previamente descrito (10).
En el estudio descriptivo de los datos las variables cualitativas se presentan con su distribución de frecuencias. Las variables cuantitativas se resumen con su media y desviación estándar (DE). Las variables cuantitativas que muestran una distribución asimétrica se resumen con la mediana y el rango intercuartílico (RIQ). En la comparación de los parámetros entre los grupos de estudio se evalúa la asociación mediante los test no paramétricos test de la U de Mann-Whitney o el test de Kruskall Wallis en función de si son dos grupos o más, respectivamente. Se usan estos test no paramétricos debido a que los grupos tienen un pequeño tamaño muestral. Para todas las pruebas se acepta un valor de significación del 5%. El procesamiento y análisis de los datos se realiza mediante el software estadístico IBM SPSS Statistics v.2º.
Todos los índices hematológicos y hallazgos clínicos fueron llevados a cabo con el consentimiento informado previo de los pacientes. Y todos los experimentos fueron realizados de acuerdo con la Declaración de Helsinki.
RESULTADOS
Setenta y tres pacientes, 33 hombres y 40 mujeres, con un rango de edad de entre 5 meses y 65 años, presentaron β-talasemia heterocigota asociada a triplicación de genes alfa globina (αααanti 3.7).
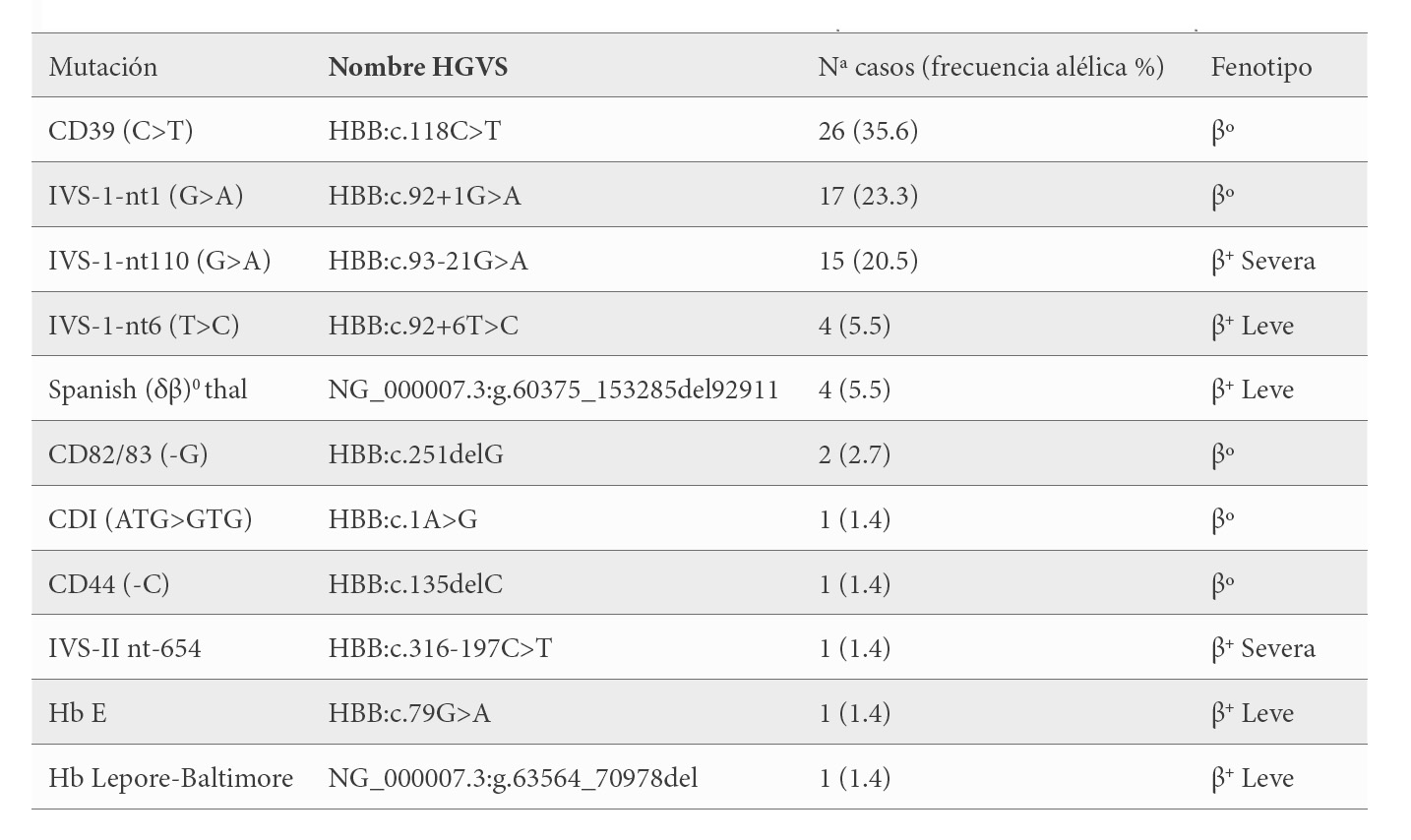
Genéticamente, todos fueron portadores de una única alteración en el gen HBB, identificándose 11 mutaciones diferentes. Estas mutaciones del gen β son: cinco β0-talasemia que agrupan al 64.4 % de los pacientes; 2 β+-talasemia severa en el 21.9 % de los casos y 4 β+-talasemia leve en 10 pacientes (13.7 %), siendo la más frecuente con un 36% la mutación de transversión de una C>T en el CD 39 del 2º exón [β39(C5) Gln>Stop; HBB:c.118C>T] seguida de la sustitución de una G>A en el nucleótido 110 del primer intrón [β nt 252 G>A; HBB:c.93-21G>A] con un 20.5% (Tabla 2).
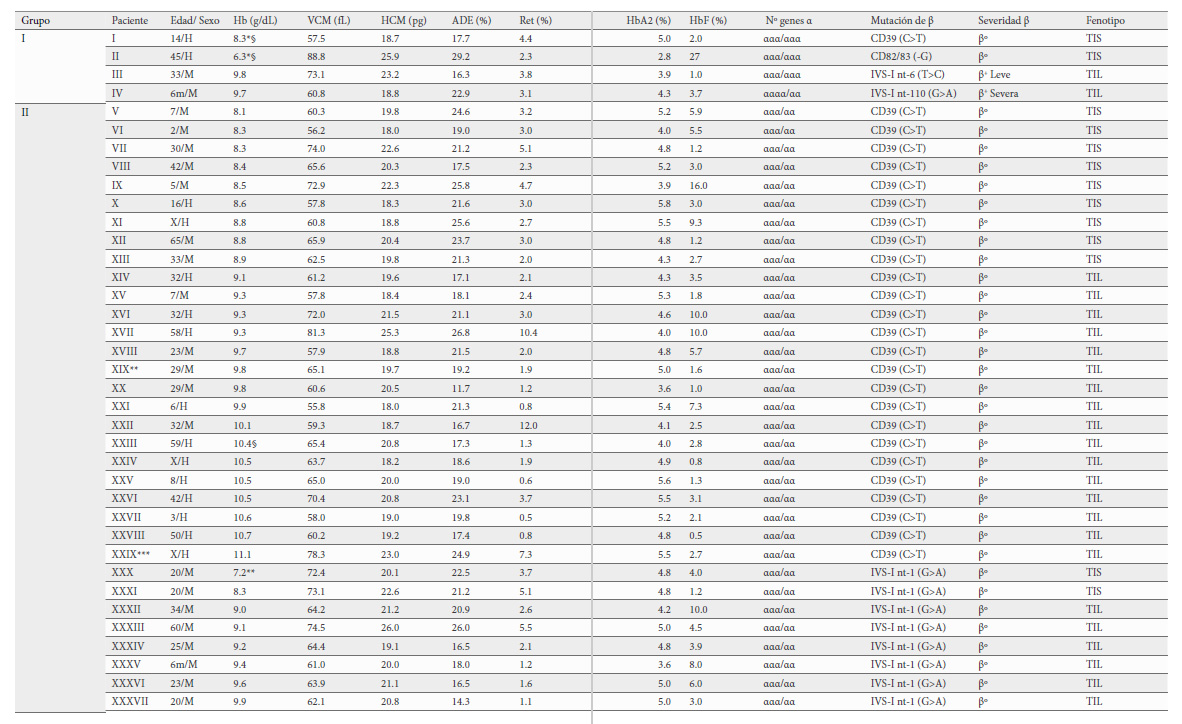
Se han clasificado los genotipos en tres grupos según el número de genes α globina y la severidad de la alteración en el gen globina. El grupo I corresponde a 4 pacientes que han coheredado 6 genes α y una mutación en el gen HBB que puede ser β0-talasemia (2 pacientes), β+-talasemia severa (1 paciente) y β+-talasemia leve (1 paciente). Los grupos II y III los constituyen pacientes con 5 genes α globina. El grupo II, está formado por 64 pacientes con β0-talasemia (45 pacientes), β+-talasemia severa (15 pacientes) o β+-talasemia leve (4 pacientes) y el grupo III con 5 pacientes portadores de mutaciones que afectan a los genes β y δ globina y que fenotípicamente son considerados como β+-talasemia leve (Tabla 3).

Aquellos que presentaron β-TI severa (TIS) mostraron entre otros síntomas clínicos debilidad, subictericia conjuntival, cálculos biliares, esplenomegalia y pérdida de peso con palidez. Mientras que los que fueron clasificados como β-TI leve (TIL) mostraron retraso del crecimiento enlentecido, algunos, pubertad retrasada y esplenomegalia y otros, problemas óseos. En los rasgos talasémicos (RT) la mayoría presentaron fatiga, sensación de mareo, dolor de cabeza y palidez de piel.
Desde el punto de vista hematológico se dice que en la β-TI los niveles de Hb se sitúan por encima de 7 g/dL. En nuestro estudio y dentro de cada grupo la diferenciación entre TIS, TIL y RT se hizo en función de estos niveles. Así, pacientes con Hb 7-9 g/dL fueron considerados como TIS; con Hb 9.1-11.5 g/dL como TIL y cuando la Hb >11.5 g/dL fueron clasificados como RT. De tal modo que distribuidos según los grupos quedarían, Grupo I: 2 TIS y 2 TIL; Grupo II: 16 TIS, 39 TIL y 9 RT y Grupo III: 5 RT.
Los valores medios de los parámetros hematológicos de los pacientes asignados a los diferentes grupos y clasificados como TIS, TIL y RT aparecen recogidos en la Tabla 4.

discusión
En el fenotipo de la talasemia intermedia intervienen diversos factores entre ellos el desequilibrio aumentado de la síntesis de α/β globina, así como la severidad de la mutación de la β-talasemia.
Nuestro laboratorio es centro de referencia en España para el estudio y diagnóstico molecular de hemoglobinopatías estructurales y talasemias, recibiendo anualmente aproximadamente 900 muestras, a todas ellas se le somete al cribado para las mutaciones más frecuentes de α-talasemia y triplicación génica (11). Desde enero de 2010 hasta diciembre de 2019 se recibieron 8870 muestras, de las cuales 73 individuos mostraron la presencia simultánea de triplicación o cuadruplicación de genes α y β-talasemia heterocigota. Es la mayor serie publicada de este tipo de asociación.
En España, la frecuencia de la β-talasemia heterocigota es de aproximadamente el 1.5 % mientras que la incidencia de la triplicación de genes alfa globina es desconocida (12). De modo que, en nuestro país, la asociación de ambas entidades está poco establecida, de ahí que nos hayamos planteado recopilar todos los casos que mostraron simultáneamente triplicación de genes α globina y β-talasemia y analizarlos desde el punto de vista hematológico y fenotípico para, comprobar si la influencia de este aumento de genes alfa en pacientes portadores de una única mutación en un locus beta globina, presenta un fenotipo de talasemia intermedia en la población española, al igual que en poblaciones de otros países ya estudiadas.
El espectro clínico de la TI es muy amplio, así como el fenotipo hematológico. La anemia, es el primer signo de sospecha y en los pacientes con β-TI puede ser de moderada a leve, con una producción de glóbulos rojos adecuada para mantener niveles de Hb > 7 g/dL y sobrevivir sin necesidad de transfusiones sanguíneas de forma regular; se recomiendan transfusiones cuando existe un descenso evidente de la cifra de Hb < 5 g/dL. Algunos son asintomáticos hasta la edad adulta o manifiestan un fenotipo más benigno. El análisis molecular sigue siendo la herramienta de diagnóstico definitiva para el fenotipo de talasemia intermedia (13,14).
Un total de 11 mutaciones diferentes entre β0-talasemia, β+-talasemia severa y β+-talasemia leve, en el gen HBB, han sido identificadas en nuestra serie, siendo las mayoritarias las β0-talasemias. Solapándose las mutaciones y el porcentaje con la frecuencia publicada en la población española (12).
Los pacientes del grupo I constituido por 4 casos, son los que tienen 6 genes alfa globina. Tres fueron homocigotos para la triplicación de genes α globina (ααα/ααα), dos de ellos con anemia severa, hiperesplenismo, osteopatía y requerimientos transfusionales y en tratamiento quelante con deferroxamina, ambos pacientes presentaron una β0-talasemia. El tercer caso, sin requerimiento transfusional fue considerado TIL, la severidad de la β-talasemia fue β+-talasemia leve. Con una clínica compatible con anemia leve, palidez, fatiga e irritabilidad. El cuarto paciente también mostró un fenotipo de TIL, y sin embargo la mutación de la β-talasemia fue severa, pero la distribución de los 6 genes alfa globina correspondió con una cuadruplicación (αααα/αα). Hay que destacar que la paciente, cuando se le realizó el estudio contaba con tan solo 6 meses de edad, pero ya tenía pérdida de peso, palidez, esplenomegalia y cabría esperar que con la edad desarrollara TIS. Por tanto, independientemente de la distribución de los 6 genes alfa globina, ya sea triplicación homocigota (ααα/ααα) o cuadruplicación heterocigota (αααα/αα), la asociación con una β-talasemia heterocigótica da como resultado una anemia de severa a moderada que puede o no requerir de terapia transfusional, siendo la severidad de la mutación del gen HBB la que determinaría la variación clínica como ya apuntaron Traeger-Synodinos et al. y Sollano et al. (15,16). Respecto a los datos hematológicos son más severos en los pacientes que muestran TIS, aunque no se han encontrado diferencias a nivel estadístico debido al bajo tamaño muestral (Tabla 4).
El grupo II fue el más numeroso (64 pacientes), todos ellos fueron portadores de cinco genes alfa globina (ααα/αα) excepto uno que junto a la triplicación en un alelo coheredó también una deleción de 3.7 kb (ααα/-α3.7). Este paciente, según los datos hematimétricos, fue clasificado como RT presentando en el gen HBB una mutación categorizada como severa [IVS-1-nt1 (G>A)], de modo que, aunque tiene una triplicación, al asociarse con la deleción 3.7 kb la carga genética de α globina es 4, por lo tanto, fenotípicamente sería portador de una mutación en el gen HBB y al tener 4 genes alfa su manifestación clínica y de los parámetros eritrocitarios sería como un rasgo talasémico beta sin complicaciones como ya apuntaran Villegas et al en el año 1997 (17).
En este grupo, la mayoría de los pacientes (39/64) mostraron un fenotipo de TIL a pesar de que el 70 % de los pacientes fueron portadores de una β0-talasemia, por lo que hubiéramos esperado una mayor proporción de pacientes con una expresión fenotípica más severa. Dieciocho presentaron esplenomegalia, 15 crecimiento lento y pubertad retrasada y 8 problemas óseos.
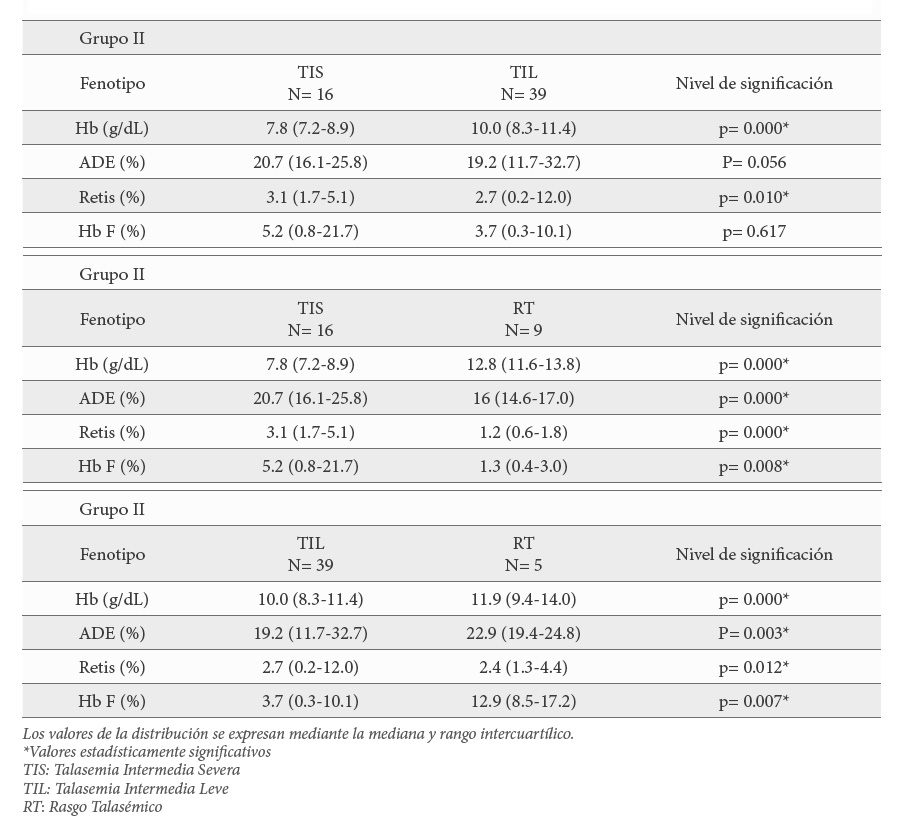
Se obtuvieron diferencias estadísticamente significativas entre pacientes de los 3 fenotipos (TIS, TIL y RT) para la cifra de hemoglobina, el ADE, en el número de reticulocitos y en el porcentaje de Hb F. Los parámetros hematimétricos más graves los presentaron los 16 pacientes con TIS, la menor tasa Hb (8.4 g/dL); el mayor porcentaje de Hb F (5.2%), así como el RDW (20.7 %) y los reticulocitos (3.1 %) más altos (Tabla 4) y que presentaron hepatoesplenomegalia, lesiones esqueléticas, retraso del crecimiento y masas paravertebrales.
El análisis posterior de estas variables, estadísticamente significativas, mediante comparaciones 2 a 2 mostró que, hubo diferencias entre los pacientes con TIS y TIL; entre pacientes con TIS y RT, así como entre TIL vs RT para todas las variables. De modo que, en todos estos parámetros, los valores más cercanos a la normalidad los presentan los pacientes con RT y los valores más extremos los pacientes con TIS (Tabla 5).
Desde el punto de vista de la severidad de la alteración de la β-talasemia se observa que el 73% de los casos con una β0-talasemia se comportan como TIL, donde no hubo lesiones hematopoyéticas extramedulares, y al no haber transfusiones periódicas no se objetivaron lesiones hepáticas ni ninguna de las complicaciones derivadas de la sobrecarga férrica. Cuando la mutación es β+-talasemia severa la mitad de los pacientes (8/15) presentan TI severa o leve, con más edad presentaron úlceras en las piernas, una de las complicaciones de la TI probablemente como consecuencia a la reducción de la oxigenación tisular que se cree que se debe a la combinación de anemia, hipercoagulabilidad y eritropoyesis ineficaz. Y cuando se corresponde con una β+-talasemia leve, en el 75% de los casos muestra TIL, frente al 25% que cursa como RT sin complicaciones. Estos resultados vendrían a enriquecer los datos que, de este tipo de asociación, previamente publicados en otros países de la cuenca mediterránea como Italia y Grecia, así como en los del cinturón talasémico como India (15,18,19,20), en donde esta asociación está bien establecida y todos coinciden en que cursan como TI ya sea severa o leve con esplenomegalia, retraso en el crecimiento y en el desarrollo hormonal, y también problemas óseos, que pueden o no necesitar terapia transfusional.
El análisis estadístico, independientemente del fenotipo, en relación con la severidad de la mutación de la β-talasemia, sólo arrojó diferencias estadísticamente significativas entre pacientes con mutaciones β0-talasemia y β+-talasemia severa, ya que el número de pacientes con mutaciones +-talasemia leve fue muy bajo. Los parámetros que resultaron ser significativos fueron la Hb (9.6 vs 11 g/dL), el ADE (19.8 vs 16.6 %) y el porcentaje de Hb F (4.3 vs 2.9 %), presentando los valores más extremos los pacientes portadores de una β0-talasemia. La cifra de hemoglobina se va reduciendo con la gravedad del fenotipo. El ADE es más elevado en aquellos pacientes con una β0-talasemia ya que es un marcador que permite ver cuán diferentes son las poblaciones eritrocitarias, y a mayor gravedad de la talasemia la anisocitosis será más marcada (ADE alto) con 2 poblaciones bien diferenciadas (eritrocitos microcíticos e hipocrómicos y eritrocitos normocíticos y normocrómicos). Y respecto a la Hb F, está aumentada en los portadores de una β0-talasemia porque al existir menor cantidad de cadena β globina disponible para la formación de dímeros αβ, el exceso de cadenas α se asociará no sólo a las cadenas δ globina para conformar la Hb A2 (α2δ2) sino también a las cadenas γ globina para el ensamblaje de Hb F, disminuyendo el efecto nocivo de la precipitación intracelular de las cadenas α no unidas. Estos resultados confirman que la TI es más grave cuando es causada por una β0-talasemia, que cuando la mutación es debida a una β+-talasemia severa. Aunque no se han encontrado diferencias significativas en los reticulocitos, a pesar de estar aumentados en todos ellos, creemos que no sólo el exceso de cadenas α contribuye a la síntesis de Hb A2 y Hb F sino que también hace que se formen tetrámeros insolubles, los cuales precipitan y dañan la membrana de los glóbulos rojos y junto a la severidad de la mutación de la β-talasemia influirían, agravando la eritropoyesis ineficaz con resultados evidentes en la clínica, coincidiendo con lo publicado por Camaschella et al. y Beris et al, en la TI (21,22).
Los análisis para ver si dentro de cada mutación pudiera haber diferencias entre las TIS y las TIL no ha mostrado significación alguna, a excepción de la alteración CD39 (C>T) que mostró que la Hb era más baja en los que presentaban TIS que los de TIL, constatándose que la cifra de hemoglobina se va reduciendo con la gravedad.
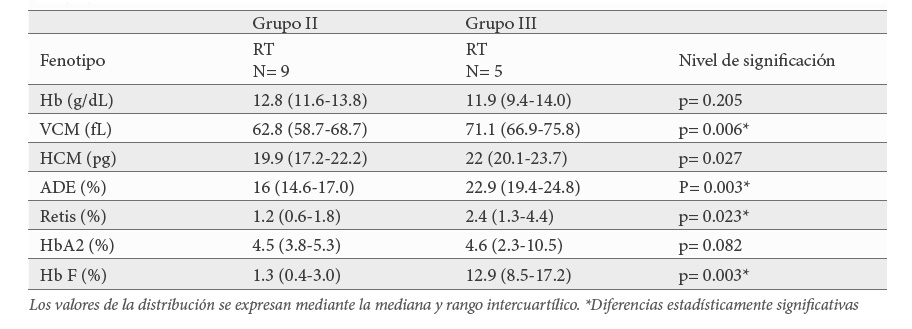
El grupo III formado por 5 pacientes, son aquellos que aparte de la triplicación de genes alfa globina han heredado una δβ-thalassemia Spanish o una Hb Lepore-Baltimore. Este tipo de alteraciones en el locus beta, donde tanto el gen δ como el β globina se ven comprometidos, se comportan fenotípicamente como una β+-talasemia leve ya que el aumento en la síntesis de Hb F contribuye a suavizar el efecto del déficit de síntesis de cadena β globina. También este aumento en los niveles de Hb F contribuiría a que los posibles efectos coadyuvantes de la triplicación de los genes alfa globina en el fenotipo serían menos exacerbados. De hecho, todos presentaron microcitosis (71.1 fL); hipocromía (22 pg); reticulocitos (2.4 %) y Hb A2 (3.1 %) normales y muy aumentados los niveles de Hb F > 8.5 % compatible con un fenotipo de RT con debilidad, cefaleas, y palidez. Este tipo de asociación ya fue descrito por Camaschella et al en Italia y Altay et al, en Irán. En Italia la coexistencia de triplicación de genes α globina en estado heterocigoto y una δβ-talasemia Sicilian o una Hb Lepore Boston se comporta como TI, mientras que en Irán este tipo de asociación al igual que en nuestra serie se describe como RT (21,23).
El último análisis estadístico consistió en ver si había diferencias importantes entre los parámetros hematimétricos de los pacientes con RT del grupo II (β+-talasemia leve) y los del grupo III (δβ-talasemia o Hb Lepore-Baltimore). Los datos mostraron diferencias en todos los parámetros, salvo en los niveles de Hb y de Hb2. Tanto el VCM, la HCM, así como el ADE se acercaron más a la normalidad en los pacientes del grupo III mientras que los reticulocitos estuvieron aumentados al igual que la Hb F (Tabla 6).
El hecho de realizar el cribado de las mutaciones más frecuentes responsables de α-talasemia y de la triplicación de genes α globina a todos los pacientes que son estudiados en nuestro laboratorio, nos ha permitido tener la mayor serie de pacientes con β-talasemia heterocigota asociada a triplicación de genes alfa globina, lo cual ha mostrado que el efecto de la asociación de una triplicación de genes α globina con una mutación β-talasemia heterocigótica es muy variable. El fenotipo puede ir desde el rasgo talasémico sin complicaciones hasta una talasemia intermedia que puede llegar a ser transfusión dependiente. Y, aunque el diagnóstico molecular de β-TI puede ser orientativo, hay que valorar en conjunto al paciente para un diagnóstico de β-TI certero. Por todo esto es muy importante el diagnóstico molecular y el consejo genético, ya que aunque la heredabilidad de una triplicación de genes alfa, es en muchos casos infradiagnosticada y no presenta complicaciones clínicas, el hecho de poder coheredarse con otra patología como es la β-talasemia heterocigota puede agravar a ésta, llegando a necesitar terapia transfusional con todas las complicaciones que este tipo de terapias conllevan así como las secuelas clínicas derivadas de la eritropoyesis ineficaz y de la anemia que se observan en las talasemias intermedias.
BIBLIOGRAFÍA
- Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. Lancet. 2012;379: 373-383
- Musallam KM, Rivella S, Vichinsky E, Rachmilewitz EA. Non-transfusion-dependent thalassemias. Haematologica. 2013;98: 833-844
- Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010;5: 11
- Vitrano A, Calvaruso G, Lai E, et al. The era of comparable life expectancy between thalassaemia major and intermedia: First time to revisit the mayor-intermedia dichotomy? Br J Haematol. 2017;176: 124-130
- Sturgeon P, Itano HA, Bergren WR. Genetic and biochemical studies of intermediate types of Cooley’s anaemia. Br J Haematol 1955;1: 264–277
- Galanello R, Cao A. Relationship between genotype and phenotype. Thalassemia intermedia. Ann NY Acad Sci 1998;850: 325–333
- Weatherall DJ, Clegg JB. The thalassaemia syndromes. Blackwell Science, Oxford. 2001
- Sankaran VG, Lettre G, Orkin SH, Hirschhorn JN. Modifier genes in Mendelian disorders: The example of hemoglobin disorders. Ann NY Acad Sci 2010;1214: 47–56
- Steinberg MH, Forget BG, Higgs DR, Weatherall DJ. Disorders of hemoglobin: Genetics, pathophysiology, and clinical management. Cambridge University Press, New York. 2009
- Torrejón MJ, Ortíz-Cabrera NV, M Nieto J et al. Hb Moncloa: A new variant of haemoglobin that interferes in the quantification of Hb A1c. Clin Biochem. 2017; 50: 521-524.
- de la Fuente-Gonzalo F, Nieto JM, Villegas A, González FA, Martínez R, Ropero P. Characterization of deletional and non-deletional alpha globin variants in a large cohort from Spain between 2009 and 2014. Ann Hematol. 2019;98: 1537-1545.
- Villegas A, Ropero P, González FA, Anguita E, Espinós D. The thalassemia syndromes: molecular characterization in the Spanish population. Hemoglobin 2001; 25: 273‐283.
- Capellini MD, Cohen A, Porter J, Taher A, Viprakasit V (eds). Guidelines for the management of transfusion dependent thalassaemia (TDT). 3a edicion. Thalassemia International Federation. 2014.
- Origa R, Baldan A, Marsella M, Borgna-Pignatti C. A complicated disease: what can be done to manage thalassemia major more effectively?. Expert Review Hematol 2015;8: 851-862
- Traeger-Synodinos J, Kanavakis E, Vrettou C, et al. The triplicated alpha-globin gene locus in beta-thalassaemia heterozygotes: clinical, haematological, biosynthetic and molecular studies. Br J Haematol 1996; 95: 467‐471.
- Sollaino MC, Paglietti ME, Perseu L, Giagu N, Loi D, Galanello R. Association of α globin gene quadruplication and heterozygous β thalassemia in patients with thalassemia intermedia. Haematologica 2009; 94: 1445‐1448.
- Villegas A, Muñoz JA, Risueño CF, et al. Association of alpha and beta thalassemia with alpha gene triplication in one family. Med Clin (Barc). 1997;108:781-783
- Camaschella C, Mazza U, Roetto A, et al. Genetic interactions in thalassemia intermedia: Analysis of β-mutations, α-genotype, γ-promoters, and β-LCR hypersensitive sites 2 and 4 in Italian patients. Am J Hematol 1995; 48: 82-87
- Theodoridou S, Balassopoulou A, Boutou E, et al. Coinheritance of Triplicated Alpha-Globin Gene and Beta-Thalassemia Mutations in Adulthood: Ten Years of Referrals in Northern Greece. J Pediatr Hematol Oncol. 2020; 42(8):e762-e764.
- Mehta PR, Upadhye DS, Sawant PM, et al. Diverse phenotypes and transfusion requirements due to interaction of β-thalassemias with triplicated α-globin genes. Ann Hematol 2015; 94(12):1953‐1958.
- Camaschella C, Kattamis AC, Petroni D, et al. Different hematological phenotypes caused by the interaction of triplicated α-globin genes and heterozygous β-thalassemia. Am J Hematol 1997; 55(2):83–88
- Beris P, Solenthaler M, Deutsch S, et al. Severe inclusion body β-thalassaemia with haemolysis in a patient double heterozygous for β0-thalassaemia and quadruplicated α-globin gene arrangement of the anti-4.2 type. Br J Haematol 1999;105(4):1074–1080
- Altay C, Öner C, Öner R, Gümrük F, Mergen H, Gürgey A. Effect of α-gene numbers on the expression of β-thalassemia intermedia, β-thalassemia and (δβ)0-thalassemia traits. Hum Hered 1998; 48(3):121-125.
DECLARACIÓN DE TRANSPARENCIA
Los autores/as de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
Paloma Ropero
Servicio de Hematología. Hospital Clínico San Carlos
C/ Profesor Martín Lagos s/n · 28040 Madrid
Tlf.: +34 913 303 321 | E-Mail: paloma.ropero@salud.madrid.org
Año 2021 · número 138 (01) · páginas 60 a 71
Enviado: 11.03.21
Revisado: 16.03.21
Aceptado: 12.04.21