

Resumen
La piropoiquilocitosis hereditaria es una membranopatía rara producida por mutaciones en la α-espectrina, que cursa con anemia hemolítica de intensidad variable. La visualización de poiquilocitos microcíticos en el frotis de sangre periférica es un hallazgo característico pero no exclusivo de esta patología. Aunque el diagnóstico definitivo se realiza mediante la identificación de mutaciones relacionadas por medio del empleo de secuenciación masiva, la citología continúa siendo una herramienta útil para la realización de un correcto diagnóstico diferencial y en la orientación del estudio molecular. Además, su papel en el seguimiento de estos pacientes permite la identificación de un subgrupo de individuos que evolucionará desde el punto de vista clínico y morfológico a una eliptocitosis congénita leve. Estos sujetos se han clasificado por algunos autores como eliptocitosis congénita común con poiquilocitosis infantil. A continuación, se expone un caso ilustrativo de este subgrupo de pacientes.
Abstract
Hereditary pyropoikilocytosis is a rare membrane disorder caused by mutations in the genes encoding alpha-spectrin. Its main manifestation is variable hemolytic anemia. Microcytic poikilocytes in peripheral blood smear are a characteristic but not exclusive finding. Although definite diagnosis is made by massive sequencing identification of the related mutations, cytology is still a useful tool for carrying out an appropriate differential diagnosis and in guiding the molecular study. In addition, morphological follow-up allows the identification of a subgroup of patients that will evolve clinically and morphologically to mild congenital elliptocytosis. These subjects have been classified by some authors as infantile poikilocytosis. We present the case of a patient with infantile transient poikilocytosis.
Palabras clave: Piropoquilocitosis hereditaria; Eliptocitosis hereditaria; Anemia; Neonato; Poiquilocitos microcíticos; Alelo Lely.
Keywords: Hereditary pyropoikilocytosis; Hereditary elliptocytosis; Anemia; Neonates; Microcytic poikilocytes; Allele alpha lely.
Las eliptocitosis hereditarias son un subgrupo de membranopatías, caracterizadas por la presencia de eliptocitos en el frotis de sangre periférica (FSP). Se producen, generalmente, por mutaciones a nivel de la α-espectrina, β-espectrina y la proteína 4.1. Estas mutaciones, aunque en su mayoría son hereditarias pueden aparecer de novo hasta en un 25% de los pacientes [1].
La piropoquilocitosis hereditaria (PPH) es un subtipo poco frecuente de eliptocitosis hereditaria, producida por mutaciones en homocigosis o doble heterocigosis en el gen que codifica la α-espectrina. Estas alteraciones condicionan modificaciones de las uniones horizontales del citoesqueleto de la membrana del hematíe, con pérdida de su módulo de elasticidad y su consecuente fragmentación. Cursa con anemia hemolítica de intensidad variable que puede presentar requerimientos transfusionales desde los primeros meses de vida. En el FSP se observan alteraciones morfológicas eritrocitarias características pero no exclusivas de esta patología, que recuerdan a las observadas en los pacientes que han sufrido quemaduras térmicas. Aunque la morfología presenta un papel importante en la realización de un correcto diagnóstico diferencial, a día de hoy, el diagnóstico definitivo se confirma mediante estudios moleculares por secuenciación masiva [1-5].
A continuación presentamos el diagnóstico y evolución de un paciente con piropoiquilocitosis infantil.
Desde el servicio de Pediatría se solicitó un FSP de un paciente con hemoglobina de 106 g/L, hematocrito 31,4%, volumen corpuscular medio (VCM) 87,3 fL, hemoglobina corpuscular media (HCM) 29,5 pg y un ancho de distribución eritrocitaria (ADE) de 47,4% al momento del nacimiento. Se había objetivado una anemización progresiva durante las primeras semanas de vida hasta alcanzar 63 g/L de hemoglobina acompañada de reticulocitos del 13%, haptoglobina de 7 mg/dL , LDH y bilirrubina total de hasta 1973 U/L y 225,72 μmol/L, respectivamente, y test de Coombs directo negativo; requiriendo de una transfusión puntual. El FSP presentaba marcada anisopoiquilocitosis con frecuentes poiquilocitos microcíticos y policromatofilia (figura 1), con algunos eritroblastos de morfología normal. El estudio mediante hematimetría y FSP de ambos progenitores no mostró alteraciones.

Se descartó el diagnóstico de talasemia mediante la amplificación de sondas dependiente de ligandos múltiples y secuenciación directa de los genes de globina por el método de Sanger. El estudio de la expresión de Eosin-5-Maleimida (EMA) demostró un detrimento de expresión del 25,22% con ratio de 0,75 respecto a la media de expresión de los controles. Por secuenciación masiva se encontró en un alelo la variante genética His469del en SPTA1, y en el otro la mutación Leuc1858Val que determina el alelo LELY (Low allele expression Lyon).
En controles posteriores realizados a los seis meses y al año se objetivó una mejoría progresiva de los niveles de hemoglobina, presentando a los dos años: hemoglobina de 122 g/L, hematocrito 37%, VCM 72 fL, HCM 23,7pg y ADE 17,4%. De forma paralela, en el FSP se observó un cambio progresivo en la morfología eritrocitaria con incremento progresivo de eliptocitos y disminución de los poiquilocitos microcíticos (figura 2).
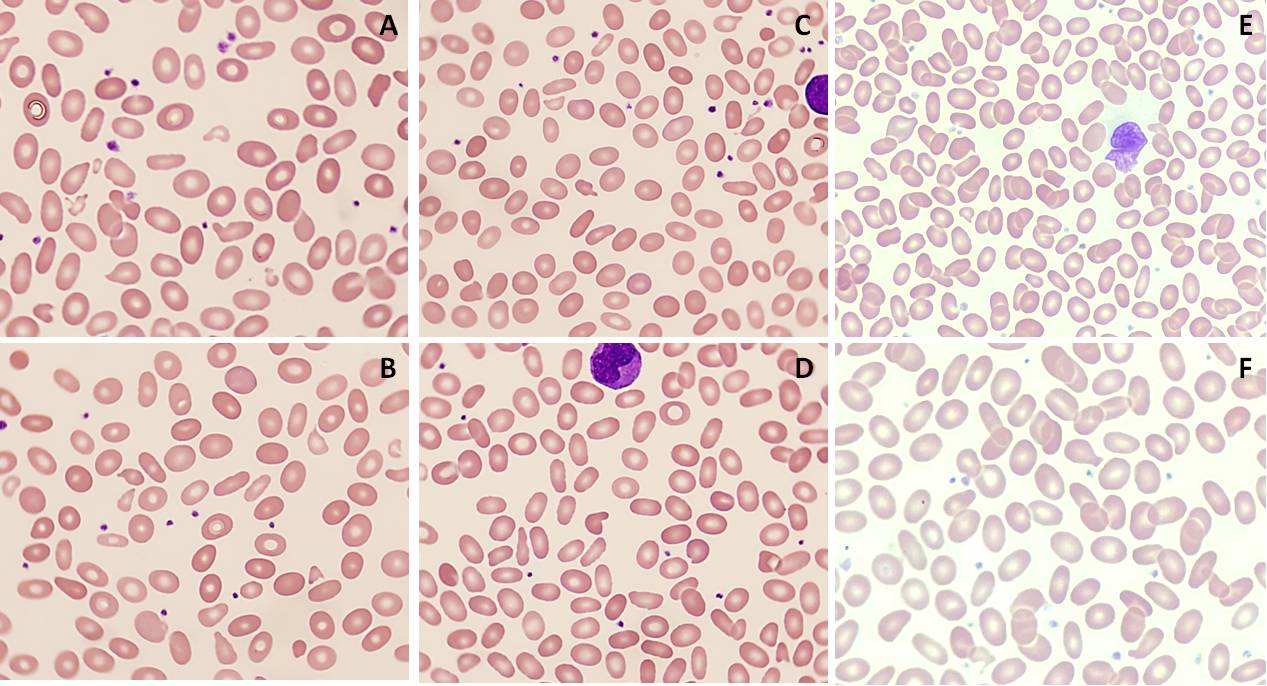
El diagnóstico de la PPH es complejo, ya que además de una clínica compatible precisa de una correcta identificación de las alteraciones morfológicas características para mantener una alta sospecha y poder, de este modo, orientar el estudio molecular. Los poiquilocitos microcíticos son hematíes de pequeño tamaño y bordes redondeados, producto de la fragmentación por gemación de los hematíes. Su formación es consecuencia de la tensión a la que son sometidos los hematíes que presentan alteraciones en la uniones de las proteínas del citoesqueleto durante su paso por la circulación sanguínea. El estudio citológico de la sangre periférica nos plantea dos retos, por un lado la correcta diferenciación entre los poiquilocitos microcíticos y otras morfologías eritrocitarias, como los esquistocitos en las microangiopatías trombóticas o los degmacitos en las hemoglobinopatías inestables. Y por otra parte, la realización de un correcto diagnóstico diferencial con aquellas patologías que pueden presentar poiquilocitos microcíticos, siendo éstas principalmente las anemias megaloblásticas y los síndromes talasémicos severos como la enfermedad de la hemoglobina H, la β-talasemia mayor o la epsilon gamma delta beta talasemia heterocigota. En este diagnóstico diferencial será crucial no solo la edad y sintomatología del paciente, sino también la presencia de otras alteraciones morfológicas asociadas (pleocariocitos, dianocitos, cuerpos de Heinz, punteado basófilo, cuerpos de Howell-Jolly, etc.). Además, el empleo del test EMA puede ser de utilidad en la orientación diagnóstica, ya que desvela una población con fluorescencia disminuida correspondiente a los hematíes fragmentados [6-8].
El diagnostico definitivo se realiza mediante la identificación de las mutaciones por medio de secuenciación masiva. Las más frecuentes se encuentran en el gen STPA1 en homocigosis o doble heterocigosis. Sin embargo, es importante destacar el papel del alelo LELY en esta patología. Este alelo, presente en un 20-30% de la población, se produce por una deleción de la zona terminal de la α-espectrina dificultando la unión de la misma con la β-espectrina. Esta alteración ofrece una ventaja adaptativa a la α-espectrina producida por el otro alelo. En pacientes con mutaciones, como en nuestro caso, la presencia del alelo LELY favorece la unión de la α-espectrina mutada, produciendo clínicamente mayor hemólisis a la esperada para un individuo heterocigoto y morfológicamente la presencia de poiquilocitos microcíticos [9-10].
Entre los individuos con diagnóstico de PPH se ha identificado un subgrupo de pacientes que presentan anemia grave al nacimiento con mejoría hematimétrica progresiva entre los cuatro meses y los dos años de edad, modificando su fenotipo desde una PPH a una eliptocitosis congénita leve, como en el caso expuesto. Estos pacientes se han clasificado clínica y morfológicamente por algunos autores como eliptocitosis congénita común con poiquilocitosis infantil. Esta evolución se ha relacionado con el contenido de 2,3 difosfoglicerato (2,3-DPG) de los hematíes.
En los neonatos el contenido libre de 2,3-DPG está aumentado a consecuencia de la baja afinidad por la hemoglobina fetal. Esta molécula debilita las uniones entre espectrina, actina y proteína 4.1 agravando el defecto y produciendo un fenotipo similar al de un paciente homocigoto. Por el contrario, a partir de los cuatro meses de edad el 2,3-DPG quedará fijado a la hemoglobina A por la que presenta alta afinidad. Esto disminuirá el contenido libre del mismo permitiendo la expresión de un fenotipo acorde al genotipo del paciente [1,4,10].
En conclusión, la PPH es una causa, aunque infrecuente, de anemia no inmune en el recién nacido. El conocimiento de esta patología nos ayudará a mantener una alta sospecha, siendo la morfología esencial para la realización de un correcto diagnóstico diferencial y en la orientación del estudio molecular del paciente. Un subgrupo de individuos con PPH presentará un curso favorable hacia lo que se ha definido por algunos autores como eliptocitosis congénita con poiquilocitosis infantil.
BIBLIOGRAFÍA
- Amador Guerrero MT, Pérez Vega S, Estrada FJ et al. Las eliptocitosis hereditarias. Correlación morfológica-molecular. Rv Mex Patol Clín. 2002; 49(4):185-202.
- Iolascon A, Andolfo I, Russo R. Advances in understanding the pathogenesis of red cell membrane disorders. Br J Haematol. 2019;187(1):13-24.
- An X, Mohandas N. Disorders of red cell membrane. Br j Haematol. 2008;141(3):367-375. doi: 10.1111/j.1365-2141.2008.07091.x.
- Da Costa L, Galimand J, Fenneteau O et al. Hereditary spherocytosis, elliptocytosis, and other red cell membrane disorders. Blood Rev. 2013;27(4):167-178.
- Prchal J, Castleberry R, Parmley R et al. Hereditary Pyropoikilocytosis and Elliptocytosis: Clinical, Laboratory, and Ultrastructural Features in Infants and Children. Pediatric Res. 1982;16(6):484-489.
- Yaish H, Christensen R, Lemons R. Neonatal nonimmune hemolytic anemia. Current Opinion Pediatr. 2017;29(1):12-19.
- Mentzer W, Iarocci T, Mohandas N, et al. Modulation of erythrocyte membrane mechanical stability by 2,3-diphosphoglycerate in the neonatal poikilocytosis/elliptocytosis syndrome. J Clin Investigation. 1987;79(3):943-949.
- Zaidi A, Buck S, Gadgeel M et al. Clinical Diagnosis of Red Cell Membrane Disorders: Comparison of Osmotic Gradient Ektacytometry and Eosin Maleimide (EMA) Fluorescence Test for Red Cell Band 3 (AE1, SLC4A1) Content for Clinical Diagnosis. Frontiers Physiol.2020.11.636. doi:10.3389/fphys.2020.00636.
- Delaunay J. The molecular basis of hereditary red cell membrane disorders. Blood Rev. 2007;21(1):1-20.
- Niss O, Chonat S, Dagaonkar N et al. Genotype-phenotype correlations in hereditary elliptocytosis and hereditary pyropoikilocytosis. Blood Cells Mol Dis. 2016;61:4-9.
DECLARACIÓN DE TRANSPARENCIA
Los autores/as de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
Silvia Escribano Serrat
Departamento de Hematología y Hemoterapia, Hospital Clínico San Carlos
Calle del Prof Martín Lagos, s/n · 28040 Madrid, España
Tlf.: +34 666 959 925 | E-Mail: Silvia.escribano.serrat@gmail.com
Año 2022 · número 139 (01) · páginas 87 a 90
Enviado: 04.04.22
Revisado: 07.04.22
Aceptado: 16.04.22