

Resumen
Las talasemias son un grupo muy heterogéneo de enfermedades hereditarias que se caracterizan por una disminución o ausencia de síntesis de una cadena de globina.
Según la cadena disminuida o ausente las talasemias se clasifican en β talasemia, α talasemia y otros tipos no tan frecuentes. La β talasemia puede ser una enfermedad grave transfusión dependiente (TDT) que requiere transfusiones programadas periódicas a lo largo de toda la vida del paciente, también hay formas intermedias menos graves, no dependientes de transfusiones (TNDT) y portadores silentes o rasgo talasémico, que son asintomáticos.
Transfusión y quelación son los 2 pilares clásicos en los que se basa el tratamiento de la TDT.
El único tratamiento curativo de la enfermedad era el trasplante alogénico de progenitores hematopoyéticos, sin embargo, no es una opción terapéutica para muchos pacientes, por la limitación para encontrar donantes HLA idénticos familiares.
En la actualidad contamos con un nuevo medicamento de terapia avanzada, de edición génica, procesado “ex-vivo” en el laboratorio, mediante la técnica de CRISPR-Cas 9, que ha sido aprobado por las Agencias reguladoras FDA y EMA para el tratamiento de la TDT. Casgevy está indicado para pacientes con TDT, a partir de 12 años, en los que siendo candidatos a un trasplante alogénico de progenitores hematopoyéticos, no es posible realizarlo por carecer de un donante HLA familiar idéntico.
En este artículo comentaremos con más detalle esta nueva opción terapéutica aprobada condicionalmente, para los pacientes con TDT que se postula como un tratamiento con potencial acción curativa de la enfermedad.Abstract
Thalassemias are a very heterogeneous group of hereditary diseases characterized by a decreased or absent synthesis of a globin chain.
According to the decreased or absent chain thalassemias are classified into β thalassemia, α thalassemia and other not so frequent types. The β-thalassemia can be a severe transfusion-dependent disease (TDT) requiring periodic scheduled transfusions throughout the patient’s life, as well as less severe forms called intermediate non-transfusion-dependent thalassemia (TNDT) and silent carriers or thalassemic trait, which are asymptomatic.
Transfusion and chelation are the two classic pillars on which the treatment of TDT is based.
The only curative treatment for the disease was allogeneic transplantation of hematopoietic progenitors, but this is not a therapeutic option for many patients, due to the limitation of finding HLA-identical family donors.
We now have a new advanced therapy drug, gene-editing, processed “ex -vivo” in the laboratory, using the CRISPR-Cas 9 technique, which has been approved by the FDA and EMA regulatory agencies for the treatment of TDT. Casgevy it is indicated for patients with TDT, aged 12 years and older, who are candidates for allogeneic transplantation of hematopoietic progenitors but are unable to undergo it due to the lack of an HLA-identical family donor.
In this article, we will discuss in more detail this new therapeutic option conditionally approved for patients with TDT that it is postulated as a treatment with potential curative action of the disease.Palabras clave: Beta talasemia; Opciones terapéuticas; Quelación; Terapia génica; Edición génica.
Keywords: Thalassemia beta; Therapeutic options; Chelation; Gene therapy; Gene editing;
Introducción
Las talasemias son un grupo muy heterogéneo, tanto molecular como clínico, de anemias hemolíticas hereditarias, que se caracterizan por una disminución o ausencia de síntesis de una o más cadenas de globina, que entran a formar parte de la hemoglobina. La cadena sintetizada en menor cuantía, es por lo general normal en su composición y en su estructura, a diferencia de lo que sucede en las hemoglobinopatías estructurales (1, 2).
Desde el punto de vista genético, según la cadena que está disminuida o ausente, las talasemias se clasifican en α, β, δβ, εγδβ, δ talasemia, o γ talasemias. Además, pueden desarrollar el fenotipo talasémico algunas hemoglobinopatías estructurales que también presentan disminución de síntesis de la cadena de globina y se expresan con microcitosis e hipocromía.
La hemoglobina normal del adulto (HbA) es un tetrámero formado por 2 cadenas α y 2 cadenas β, de ahí que las talasemias más frecuentes pertenezcan a estos dos grupos.
Por otra parte, las mutaciones causantes de β o α talasemia se clasifican en βº o αº cuando el alelo alterado no da lugar a la producción de proteína y en β+ o α+ cuando hay disminución de síntesis, pero el alelo alterado aún codifica algo de cadena (3).
Desde el punto de vista clínico las β talasemias pueden clasificarse en talasemia mayor que constituye la forma más grave de la enfermedad en la cual, los pacientes dependen de transfusiones a lo largo de toda la vida, formas intermedias, menos graves y talasemia minor o rasgo talasémico, que corresponde a los portadores heterocigotos asintomáticos, con microcitosis e hipocromía.
En los últimos años existe la tendencia clínica a simplificar la clasificación de los casos sintomáticos, según los requerimientos transfusionales en talasemias dependientes de transfusiones (TDT) que para mantener su hemoglobina requieren transfusiones periódicas regulares y talasemias no dependientes de transfusiones (TNDT) que no requieren este régimen de transfusiones periódicas o incluso estos pacientes nunca se han transfundido o solamente en contadas situaciones (2, 4).
A pesar de ser una enfermedad altamente prevalente en países de la cuenca mediterránea como Italia, Chipre o Grecia, así como en otros países del sudeste asiático, norte de África o África tropical, en España la incidencia de talasemia es baja 0.1-2% de formas leves o heterocigotas (5). En los últimos 20 meses el registro Español de Eritropatología de la Sociedad Española de Hematología y Hemoterapia ha recogido 72 casos de TDT y 69 pacientes de TNTD (6,7). El estudio se ha llevado a cabo analizando los pacientes según el número de transfusiones realizadas al año, edad, procedencia familiar, datos clínicos y tratamiento, así como el estudio molecular completo de los gene β y α.
Fisiopatología
En la fisiopatología juega un papel fundamental la ausencia de cadenas beta y el exceso de las cadenas alfa sobrantes, con las que se aparea, que son muy inestables y precipitan en el interior de eritroblastos y hematíes (2, 8). Las cadenas alfa forman agregados que oxidan membranas y proteínas y que precipitan e inducen apoptosis y muerte precoz de eritroblastos en médula ósea, antes de convertirse en hematíes, originando eritropoyesis ineficaz. En la sangre periférica los hematíes son destruidos en las sinusoides esplénicas produciendo anemia hemolítica.
Como consecuencia aparece anemia crónica grave, con hipoxia, expansión de la médula ósea con múltiples alteraciones esqueléticas, hepatoesplenomegalia y eritropoyesis extramedular (1,2,3,8).
La hipoxia y el aumento de eritropoyetina, incrementan la producción de eritroferrona, con bloqueo de hepcidina y aumento de absorción de hierro a nivel intestinal, que unido a la sobrecarga férrica producida por la terapia transfusional periódica condicionan un estado de sobrecarga de hierro con daño multiorgánico. Esta sobrecarga puede afectar a hígado con fibrosis, cirrosis y en ocasiones carcinoma hepatocelular, alteración cardiaca con miocardiopatía dilatada, afectación endocrina con diabetes mellitus, hipoparatiroidismo, hipotiroidismo, e hipopituitarismo.
A larga pueden observarse otras manifestaciones clínicas como hiperesplenismo, colelitiasis, osteoporosis, úlceras maleolares e hipertensión pulmonar.
Tratamiento
El tratamiento convencional se sustenta en dos pilares fundamentales. La corrección de la anemia mediante soporte transfusional periódico y la terapéutica quelante para la prevención y tratamiento de la sobrecarga férrica.
Este tratamiento combinado ha permitido mejorar sustancialmente la supervivencia de estos pacientes, ya que sin transfusiones fallecían en la primera década de la vida y sin quelación alrededor de la segunda década.
La decisión de iniciar el soporte transfusional con concentrado de hematíes se basa en datos clínicos y la cifra de hemoglobina (Hb). Con cifras de Hb <7 g/dl se debe comenzar la transfusión. Puede programarse con cifras >7g dl, si los niños presentan retraso del crecimiento, manifestaciones óseas o esplenomegalia (4).
El régimen transfusional comienza aproximadamente entre el primer y segundo año de vida y dependiendo de los factores previamente señalados no debe retrasarse dado que el problema de aloinmunización es menor en los primeros años de vida.
Con el régimen transfusional disminuye la eritropoyesis ineficaz, se previenen las lesiones esqueléticas, la hepatoesplenomegalia y la absorción intestinal de hierro.
Antes de comenzar las transfusiones se recomienda realizar el fenotipo extendido de los antígenos eritrocitarios y transfundir concentrados de hematíes isogrupo para los antígenos del sistema ABO, Rh, Kell para evitar posteriormente aloinmunizaciones y reacciones hemolíticas (9, 10).
Se recomienda transfundir con niveles previos de Hb de 9-10 g/dl y no sobrepasando una cifra posterior de Hb de 14-15 g/dl. En general se requieren 1-2 concentrados de hematíes mensuales.
El segundo pilar importante del tratamiento es la quelación de la sobrecarga férrica.
La sobrecarga férrica es una consecuencia inevitable de la terapia transfusional crónica, de tal modo que al cabo de 4 años el paciente puede tener una carga de hierro de 20-25 g, dado que no existe un mecanismo fisiológico de excreción de hierro y las pérdidas fisiológicas son de 1mg/día.
Para eliminar el daño multiorgánico es necesario comenzar con el tratamiento quelante, aproximadamente un año después del comienzo de la terapia transfusional periódica, cuando los pacientes han recibido entre 10-20 transfusiones, o presentan una ferritina >1000 ng/ml o una concentración hepática de hierro >3mg Fe/g seco de tejido hepático. Con el tratamiento quelante ha aumentado la expectativa de vida de los pacientes. Los pacientes bien quelados tienen una supervivencia más larga que los pobremente quelados, cuya supervivencia está muy disminuida (1,2,8,11).
Se debe de evitar que los pacientes presenten valores de hierro hepático ≥15 mg/g y de ferritina ≥2500 ng/ml, ya que por encima de estos valores existe alta probabilidad de arritmias, fallo cardiaco y muerte precoz (11).
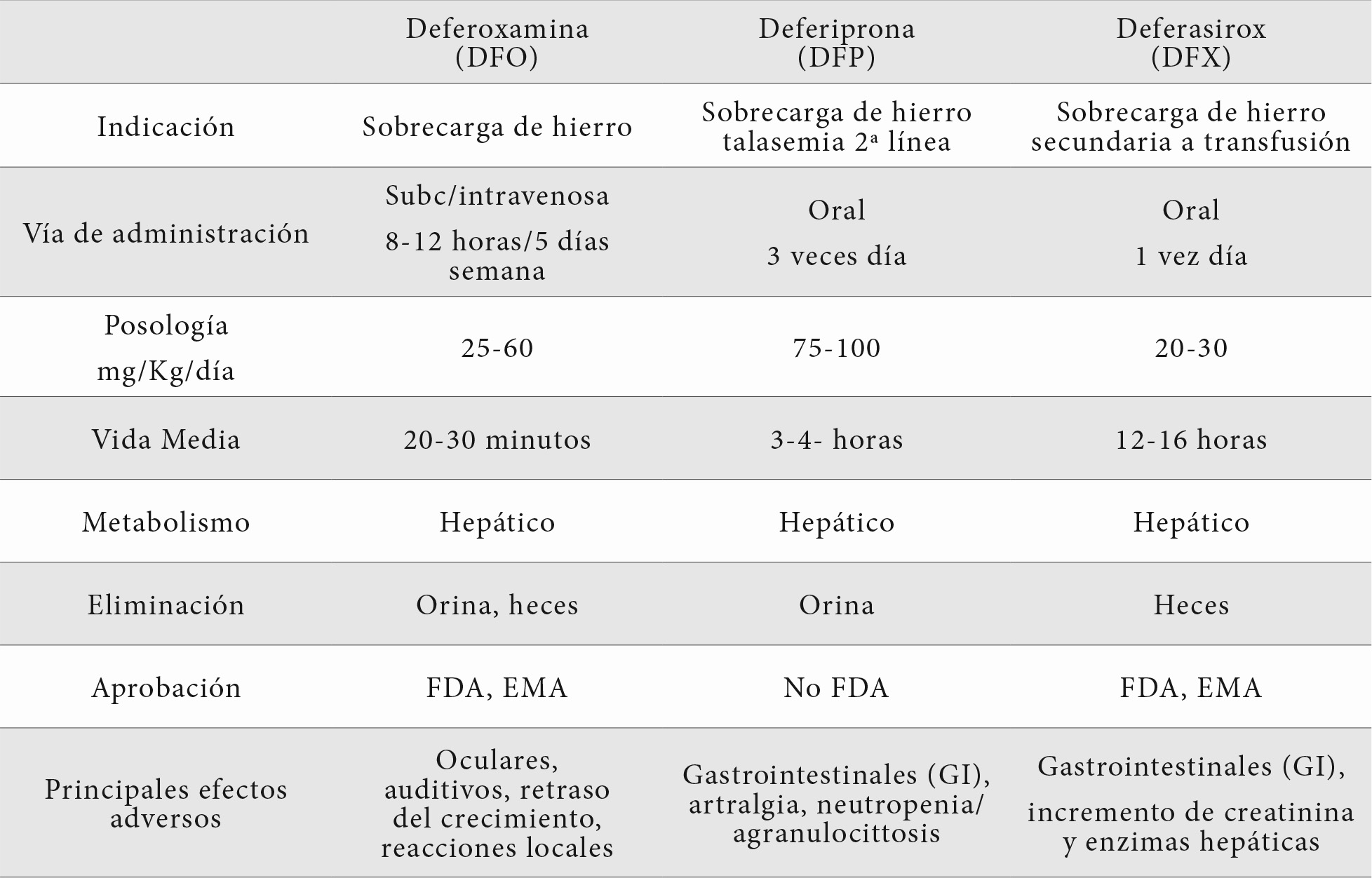
Tres medicamentos están indicados para la quelación del hierro: deferoxamina, deferiprona y deferasirox. La deferoxamina ha sido el primer quelante desarrollado que ha permitido que muchos pacientes talasémicos llegaran a una edad adulta con una buena adherencia al tratamiento. Debido a su corta vida media se ha utilizado en bombas de infusión subcutánea durante 8-12 horas y 5 días a la semana. También es de gran utilidad por vía endovenosa.
En la tabla 1 se especifican las características de estos 3 quelantes. En la actualidad el más utilizado es el deferasirox, de admiración oral y solo con una toma diaria.
La esplenectomía en el momento actual solo se prescribe ante situaciones puntuales, como hiperesplenismo o aumento importante de las necesidades transfusionales, por los problemas que pueden presentarse posteriormente, como hipertensión pulmonar o fenómenos tromboembólicos.
Hasta ahora la única terapéutica curativa de la talasemia mayor había sido el trasplante alogénico de progenitores hematopoyéticos, cuya realización ha sido muy limitada por la falta de donantes familiares HLA idénticos. Menos del 25% de los pacientes tienen un donante familiar y además se asocia con complicaciones asociadas al trasplante como rechazo del injerto y la necesidad de utilizar terapia inmunosupresora postransplante para prevenir y tratar la enfermedad injerto frente a receptor. C Li y colaboradores (12) recogen la evolución de 1110 pacientes talasémicos trasplantados y concluyen señalando que el trasplante es una buena opción si se efectúa en menores de 6 años y con una supervivencia superior al 90% si se realiza de familiar HLA idéntico. También la respuesta es buena utilizando donantes idénticos no familiares.
El trasplante haploidentico con ciclofosfamida postranplante también está obteniendo resultados muy prometedores.
Nuevos tratamientos
Con el objetivo de disminuir el número de transfusiones y las complicaciones de la enfermedad se ha intentado sintetizar nuevas moléculas, unas estimuladoras de la eritropoyesis como mitapivat o análogos de la eritropoyetina, otras estimuladores de la diferenciación celular como luspatercept, o modificadoras del metabolismo del hierro, tales como análogos de la hepcidina, inhibidores de TMPRSS6, Inhibidores de ferroportina y otros varios (13). Sin embargo, el único tratamiento que ha sido aprobado por las agencias reguladoras de USA, FDA (Food and Drug Administration) y Europa, EMA (European Medical Agency) es el luspatercept que está comercializado y por lo tanto disponible en nuestro país (14).
Luspatercept es una proteína de fusión recombinante que se une a los superligandos del factor de crecimiento TGF β y bloquea la vía SMAD 2/3 reduciendo la fosforilización y promoviendo la diferenciación eritroide en la fase tardía de la maduración medular (14,15).
En 66 Congreso de la SEHH, en octubre de 2024 el Grupo Español de Eritropatología de la SEHH ha comunicado los resultados de 15 pacientes con β talasemia TDT tratados con luspatercept con un incremento de Hb en 5 de los 15 pacientes tratados (33,3%) y en uno de ellos con independencia transfusional (16).
Terapia génica
Durante décadas se había postulado que por ser la talasemia una enfermedad monogénica, la terapia génica estaba a punto de llegar. Finalmente podemos decir que en el momento actual es una realidad.
La terapia génica tiene el objetivo de curar potencialmente la enfermedad, bien insertando un gen HBB normal o manipulado, o bien modificando alguna proteína indispensable para el buen funcionamiento de la cadena de globina deficitaria.
La terapia génica en la TDT se puede realizar mediante diferentes mecanismos ex-vivo, tras extraer las células madre progenitoras del paciente y manipularse en el laboratorio mediante diferentes procedimientos tales como lentivirus, nucleasas en dedos de zinc o la técnica de edición genética con CRISPR-Cas9. Y tras un acondicionamiento mieloablativo, posteriormente se realiza la autotransfusión de las células madre hematopoyéticas del paciente procesadas en el laboratorio, en las cuales se ha corregido la mutación genética o el defecto ocasionado por esta mutación.
Hasta ahora hay dos procedimientos de terapia génica para la TDT. Ambas terapias han sido aprobadas, por las Agencias Reguladoras Americana (FDA) y Europea (EMA) y se definen como terapia de adicción y terapia de edición (17). La primera betibeglogene autotemcel, beti- cel o zynteglo utiliza el vector lentivirus (lentiglobin BB 2095) aprobada por la EMA en 2019 y añade un gen HBB modificado, y la segunda exagamglogene autotemcel, exa-cel o casgevy utiliza la técnica CRISPR-Cas9 y será a la cual nos referimos en este trabajo más extensamente. El zynteglo ha sido retirado en Europa por decisión de la propia compañía responsable de su investigación y comercialización.
Exa-cel se basa en un hecho ampliamente conocido por los hematólogos (eritropatólogos). En la persistencia hereditaria de hemoglobina fetal (PHHF) se produce una mutación o deleción en el promotor de las cadenas gamma A o gamma G del gen HBB y como consecuencia en el periodo postnatal no se produce el cambio de Hb Fetal (HbF;2α, 2λ) a Hb A normal (2α, 2β) y los pacientes siguen manteniendo niveles elevados de HbF a lo largo de toda su vida. Estos pacientes desde el punto de vista clínico son prácticamente normales. Una Hb F > 30% mejora la enfermedad al aparearse la cadena α con la λ y por lo tanto desaparecer el desequilibrio α/β y disminuir el número de cadenas α libres. Recordemos nuevamente que la fisiopatología de la enfermedad y la gravedad de la TDT están en relación directa con el número de cadenas α sobrantes que conllevan eritropoyesis ineficaz y anemia hemolítica.
Con la técnica CRISPR-Cas9 se rompe la unión GATA1, con el gen BCL11A, que es el responsable de la represión de la cadena λ en el periodo neonatal, y se vuelve a activar nuevamente produciendo Hb F.
Exa-cel está indicado en el tratamiento de pacientes con TDT con edad superior a 12 años, en los que está indicado el trasplante de progenitores hematopoyéticos de familiar histocompatible, pero no es posible realizarlo porque no se encuentra un donante adecuado (18).
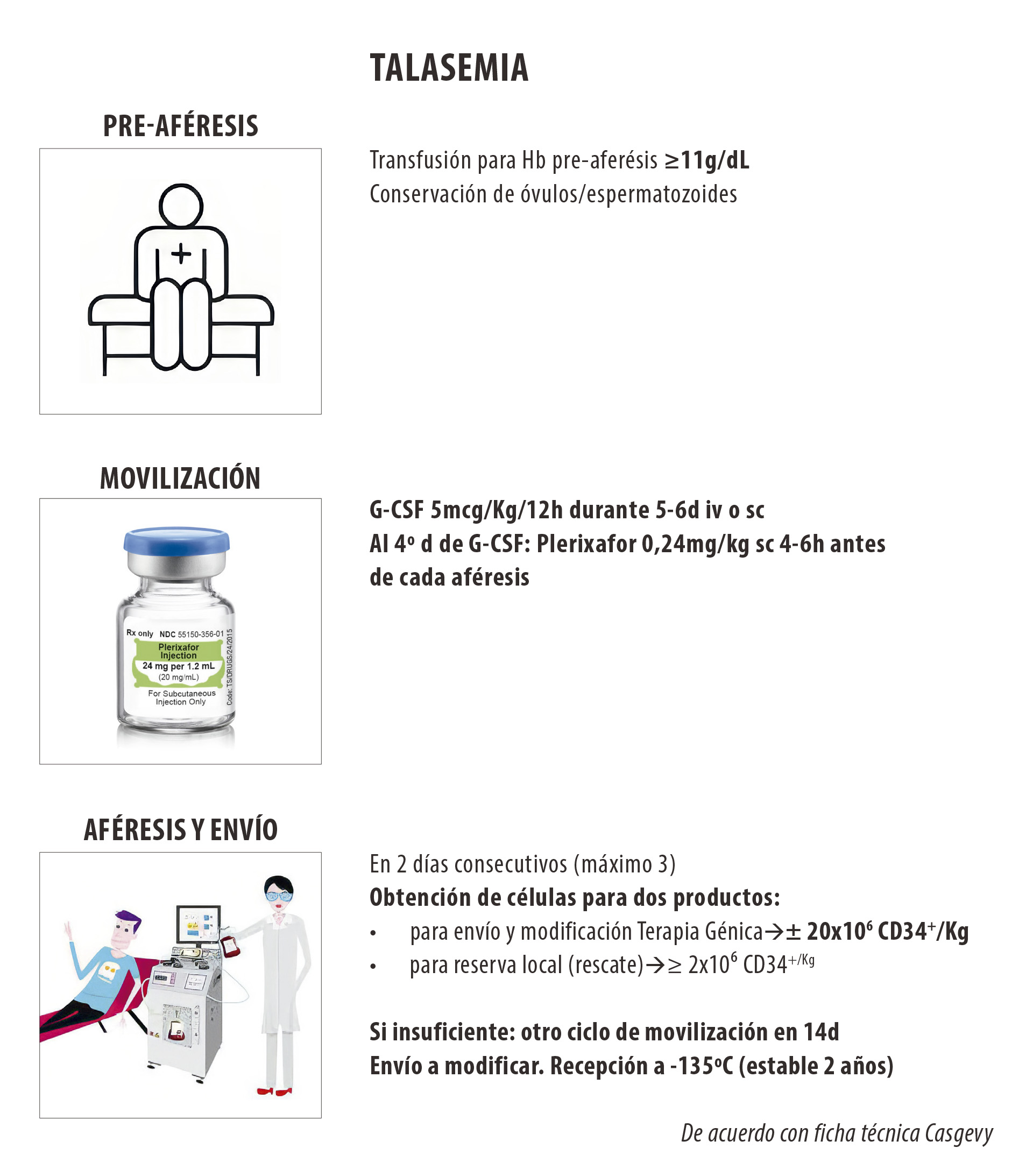
La eficacia y seguridad de exa-cel se realizó gracias a un estudio abierto, multicéntrico, de un solo brazo (ensayo CLIMB THAL- 111), en pacientes con TDT en edades comprendidas entre 12 y 35 años. El primer paso consistió en la extracción de células madre progenitoras CD 34+, tras la movilización con G-CSF y plerixafor (figura 1), envío al laboratorio para su manipulación con la técnica CRISPR-Cas9 y a continuación una vez recibidas las células manipuladas realizar un autotransplante de progenitores hematopoyéticos. La mieloablación se lleva a cabo con busulfán y a las 2 a 7 días de este tratamiento, se realiza la infusión de las células procesadas (18,19,20).
Por lo tanto, la terapia génica se basa en la infusión de las propias células del paciente y de ese modo se evitan los problemas inmunológicos derivados del transplante alogénico.
La variable principal del ensayo consistió en evaluar el número de pacientes que consiguieron la independencia transfusional tras 12 meses o más de seguimiento, manteniendo una Hb ≥ 9 g/dl (18). Se evaluaron 42 de los 52 pacientes que entraron en el ensayo y 39 cumplieron el objetivo principal (92,9%). Así mismo se cumplieron los objetivos secundarios (incremento de las Hbs total y Hb F) y a los 6 meses la Hb total ascendía a 12,2 g/dl y la Hb F era de 10,9 g/dl, de distribución pancelular y a los 24 meses persistía el incremento con cifras de Hb total de 13,1g/dl y de Hb F de 11,9 g/dl (19,20). Un año más tarde se volvieron a evaluar los 52 pacientes del estudio y en 49/52 se había conseguido la independencia transfusional, mantenida más allá de 12 meses (Hb ≥9 g/dl, respuesta del 94,2%) con una mediana de duración de la independencia transfusional de 31 meses (12,8-59,4 meses). Así mismo se cumplieron las variables secundarias con el incremento de las Hb total y Hb F.
Dentro de los efectos adversos prácticamente la totalidad de los mismos fueron atribuidos al acondicionamiento mieloablativo con busufán. Solamente 2 pacientes tuvieron manifestaciones clínicas atribuidas a exa-cel: 1 caso con fracaso del injerto y trombopenia y otro con linfohistiocitosis hemofagocítica. Ningún paciente falleció o tuvo que interrumpir el estudio por efectos adversos.
En el momento actual hay una plétora de nuevos medicamentos que evalúan tanto la terapia de adición génica como de edición genética.
La EMA ha aprobado condicionalmente Casgevy para pacientes con 12 años o a partir de esa edad, candidatos al transplante alogénico de progenitores hematopoyéticos de un familiar HLA idéntico pero que carecen del donante familiar adecuado. La designación del medicamento se ha desarrollado bajo el Programa PRIME y ha recibido la aprobación frente a una necesidad de tratamiento no cubierta.
La aprobación condicional de la EMA se ha basado en los problemas que pueden plantearse a largo plazo derivadas del escaso número de pacientes tratados hasta la actualidad y al corto seguimiento de su evolución (19). El tratamiento con exa-cel también ha sido aprobado para pacientes con enfermedad de células falciformes grave (ECF), candidatos a un transplante alogénico de donante familiar HLA idéntico pero que no tienen un donante adecuado (19,20).
La técnica de CRISPR-Cas9 requiere una guía de ARN, con la posibilidad de desarrollar aberraciones cromosómicas y necroptisis y, consecuentemente la inducción de procesos malignos. Por otra parte Cas9 rompe las dos cadenas de ADN que posteriormente necesitan ser reparadas. La reparación puede originar inserciones o deleciones que pueden ocasionar apoptosis, reordenamientos genómicos e inducir cambios neoplásicos, por lo que es necesario un seguimiento a largo plazo para descartar cualquier proceso oncogénico. En la actualidad se está investigando con la terapia de edición de bases, tales como adenina o citosina y Cas9 inactivada, que no producen rotura de las dos cadenas de ADN y por lo tanto no generan los cambios genéticos señalados.
De momento no se han observado enfermedades neoplásicas en los pacientes tratados con exa-cel, aunque no se conoce a medio y largo plazo cual puede ser el riesgo de enfermedades malignas debidas a la edición génica (17,19).
Conclusión
La talasemia transfusión dependiente es una enfermedad grave en la que el único tratamiento curativo es el transplante alogénico de progenitores hematopóyeticos, pero su realización es limitada por la falta de donantes HLA idénticos compatibles familiares y por las complicaciones asociadas al transplante como fallo del injerto, enfermedad injerto frente al receptor y la necesidad del empleo postransplante de tratamiento inmunosupresor. De ahí que la terapia génica de edición en la TDT sea una opción terapéutica muy relevante en el momento actual, con la posibilidad de la potencial curación del enfermo.
Agradecimientos
Los autores desean agradecer al Grupo Español de Eritropatología de la SEHH su inestimable ayuda en el estudio de la talasemia en España. Así como también a todos los pacientes y a sus familiares con TDT.
DECLARACIÓN DE TRANSPARENCIA
Los autores/as de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
Bibliografía
- ↑Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassemia. Lancet 2012; 379(9813): 373-383.
- ↑Taher AT, Musallan KM, Capellini MD. β-thalasemias. N Eng J Med 2021; 384(8): 727-743.
- ↑Thein SL, Molecular Bassis of β-thalasemia. Cold Harb Perspect Med 2013; 1: 3(5): 1-24.
- ↑Ropero Gradilla P, González Fernández FA. Talasemia en Eritropatología. SEHH. Ambos Marketing Services S.L. 2º edición. Barcelona 2024 ISBN. 978-84-125 780-7-2.
- ↑Villegas A, Ropero P, González FA, Anguita E, Espinós D. The thalassemia syndromes: Molecular characterization in the Spanish population. Hemoglobin 2001; 25(3): 273-283.
- ↑Menor Gómez M, Ropero Gradilla P, Fonte Leal C. et al. Registry of transfusion dependent beta-thalassemia (TDT), cases in Spain molecurlarly characterized over the past 20 months. Blood 2024; 144(supl 1): 5267.
- ↑Ropero Gradilla P, Menor Gómez M, Fonte Leal C. et al. Caraterización molecular de la talasemia transfusión dependiente (TDT) en España. Sangre 2024; 43(supl 1): 77.
- ↑Taher AT, Weatherall DJ, Capellini MD. Thalassaemia. Lancet 2018; 13: 391(10116): 155-167. Doi: 10.1016/so 140-6736(17) 31822-6.
- ↑Shah F, Sayin F, Trompeter S, Drasar E, Pig A. Challenges of blood transfusions in β-thalasemia. Blood Rev 2019; 37 (100588): 1-13. Doi.org/10.1016.
- ↑Lal A, Wong TE, Andrews J et al. Transfusions practices and complications in thalassemia. Transfusion 2018; 9999: 1-10.
- ↑Borna-Pignatti C, Capellini MD, De Sefano P et al. Cardiac morbility and mortality in deferoxamine-or deferiprone-treated patients with thalassemia major. Blood 2006; 107: 3733-3737.
- ↑Li C, Mathews V, Kim S et al. Related and urelated donor transplantion for β-Thalasemia major: results an international Survey Blood Adv 2019; 3(17): 2562-2670. Doi 101182/bloodadvances.2019000291.
- ↑Taher AT, Bou-Fakhredin R, Kattamis A, Viprakasit V, Capellini MD. Improving outcomes and quality of life for patients with transfusión-dependient β-thalassemia recomendations for best clinical practice and the use of novel strategies. Expert Rev Hematol. 2021;14(10):897-909
- ↑Capellini MD, Viprakasit V, Taher AT et al. A phase 3 trial of Luspatercept in patients with transfusion-dependent β thalassemia. N Engl J. Med. 2020; 382(13): 1219-1231.
- ↑Informe de posicionamiento terapéutico de Luspatercept (Reblozyl) en anemia dependiente de transfusiones asociada a síndromes mielodisplásicos o beta talasemia. IPT 103/2023.V1. 14 de febrero 2023.
- ↑De la Iglesia S, Morado M, Vicente A et al. Tratamiento con Luspatercept en pacientes con talasemia transfusión dependiente. Experiencia del grupo de Eritropatología. Sangre. 2024;43 (supl 1):78.
- ↑Lidonnici MR, Scaramuzza S, Ferrari G. Gene Therapy for hemoglobinopathies. Hum Gene Therapy. 2023;34(17,18) 793807- Doi 10.1089/hum. 2023. 138.
- ↑Locatelli F, Lang F, Vall D. Exagamglone Autotemcel for transfusión-depedent β thalassemia. N Engl. J. Med. 2024; 390(18): 1663-1676.
- ↑EPAR de exagamglogene autotemcel disponible en: https://www.ema.europa.eu/en/documents/product-information/exagamglogene autotemcel-epar-public-assessment-report_en pdf
- ↑Ficha técnica exaganglogén autotemcel (casgevy). Disponible en: https://www.ema.europa.eu/es/documents/product-information/ -epar- casgevy product-information
ranm tv
Ana Villegas Martínez
Real Academia Nacional de Medicina de España
C/ Arrieta, 12 · 28013 Madrid
Tlf.: +34 91 159 47 34 | E-Mail: anamaria.villegas@salud.madrid.org
Año 2024 · número 141 (03) · páginas 241 a 247
Enviado*: 12.11.24
Revisado: 20.11.24
Aceptado: 04.12.24
* Fecha de lectura en la RANM