

Resumen
Las eritrocitosis se definen como el aumento de la masa eritrocitaria por encima del 25% de su valor normal. Este incremento va paralelo al aumento del valor hematocrito.
Se conocía que en situaciones de hipoxia tisular, la eritropoyetina era el mayor estímulo para la síntesis de eritrocitos, pero no se sabía cómo las células detectaban estos cambios y se adaptaban a la disponibilidad del oxígeno. Las investigaciones realizadas por Gregg Semenza, William Kaelin y Peter Ratcliffe acerca de cómo las células perciben y se adaptan a los cambios de oxígeno, les ha merecido en el año 2019 el premio Nobel de Fisiología y Medicina. Describen cómo se lleva a cabo la regulación a nivel celular mediante la identificación de 3 proteínas, el factor inductible por hipoxia (HIF), la proteína prolin hidrolasa (PHD) y el factor supresor de tumores Von Hippel-Lindau (VHL). Estos autores analizan el mecanismo de la homeostasis del oxígeno en situaciones con aporte normal de oxígeno y en situaciones de hipoxia y por lo tanto cómo se realiza la síntesis de eritropoyetina a través del elemento respondedor a la hipoxia (HRE), localizado en la región promotora del gen de la EPO, en las células intersticiales renales peritubulares.
Clasificamos las eritrocitosis en primarias y secundarias y congénitas y adquiridas. Posteriormente se establece el diagnóstico diferencial entre los subtipos de eritrocitosis, iniciando la descripción con los criterios diagnósticos de la policitemia vera.
Se evalúan las pruebas diagnósticas necesarias para realizar su diferenciación junto con el algoritmo aplicable para programar el estudio de las eritrocitosis.
Finalmente exponemos nuestra experiencia sobre hemoglobinopatías con alta afinidad por el oxígeno, estudiadas en el Hospital Clínico San Carlos, pertenecientes a 34 pacientes de 18 familias con 11 variantes diferentes de hemoglobinopatías. Dos de ellas solamente han sido descritas en nuestro país.
A pesar de tener valores elevados de valor hematocrito y hemoglobina y en dos de ellos asociarse con talasemia y presentar nulos valores de hemoglobina A normal, los pacientes están asintomáticos.
Abstract
Erythrocytosis is defined as the increase in red cell mass above 25% of its normal value. This increase have a parallel increase of hematocrit level.
It was known that in situations of tissue hypoxia erythropoietin was the largest ensurer for erythrocyte synthesis, but it was not known how cells detected these changes and contributed to oxygen availability. The research on oxygen changes of Gregg Semenza, Willian Kaelin and Peter Radcliffe have earned them the 2019 Nobel Price in Physiology and Medicine. They describe how regulations is carried out at the cellular level by identifying 3 proteins, the hypoxia-inductible factor (HIF), prolyl hidrolase domain (PHD) and tumor suppressor factor, Von Hippel-Lindau (VHL).These authors analyze the mechanism of oxygen homeostasis in situations under well-oxygenated conditions and in situations of hypoxia and therefore as erythropoietin synthesis is performed through the hypoxia-responder element (HRE) located in region promoter of the EPO gene in peritubular renal interstitial cells.
Erythrocytosis can be classified either as primary or secondary and can be either congenital and acquired.
Subsequently the differential diagnosis is established between the subtypes of erythrocytosis, initiating the description with the diagnostic criteria of plycythemia vera.
The diagnostic test needed to perform their differentiation are evaluated along with the applicable flow chart to schedule the study of erythrocytosis.
Finally we expose our experience in hemoglobinopathies with high affinity for oxygen studied at the Clinico San Carlos Hospital, belonging to 34 patients from 18 families with 11 different variants of hemoglobinophaties. Two of them have only been described in our country. Despite having high hematocrit and hemoglobin levels and in two of then associated with thalassemia and presenting null values of normal hemoglobin A, patients are asmptomatic
Palabras clave: Eritrocitosis; Homeostasis del oxígeno; Hemoglobinopatías con alta afinidad por el oxígeno.
Keywords: Erythrocytosis; Hypoxia-inductible factors; Hemoglobinopathies; High oxygen affinity.
INTRODUCCIÓN
La eritrocitosis se define como el incremento de la masa eritrocitaria (ME) superior al 25% de su valor normal en relación con la edad, sexo y altitud de residencia. Este exceso de hematíes camina paralelo con el incremento del valor hematocrito.
Dadas las dificultades, en el momento actual, para realizar la masa eritrocitaria mediante cromo 51 y el volumen plasmático con Iodo 125, en la práctica asistencial nos referimos a eritrocitosis cuando el valor hematocrito (VH) es superior al 52% en varones y 48% en mujeres, y la hemoglobina (Hb) superior a 18.5 g/dl en varones y 16.5 g/dl en mujeres. Estos valores se acompañan siempre de una elevada masa eritrocitaria (1), sin embargo en casos límite, la determinación por métodos isotópicos puede ser de suma utilidad (2).
Se prefiere utilizar el término de eritrocitosis en detrimento de poliglobulia, reservando el nombre de policitemia vera para la enfermedad clonal, encuadrada dentro de los síndromes mieloproliferativos crónicos. Philadelphia negativos (2).
Al lado de la eritrocitosis absoluta que acabamos de definir, la eritrocitosis relativa hace referencia a un grupo de sujetos cuyo VH se encuentra elevado, pero no existe aumento de masa eritrocitaria, sino una disminución del volumen plasmático (3) por pérdida de líquido intravascular y consiguiente hemoconcentración. Es debido a diferentes causas como diarrea severa, vómitos, quemaduras, etc.
El síndrome de Gaisböck es una eritrocitosis relativa denominada también pseudopoliglobulia o poliglobulia de estrés, en pacientes generalmente fumadores, obesos, estresados, hipertensos y con hiperlipidemia e hiperglucemia. Mejoran con dieta adecuada, ejercicio físico, control de la ansiedad y el abandono del tabaco.
En esta revisión solamente nos vamos a referir a las eritrocitosis absolutas. El objetivo de este trabajo es evaluar las causas de eritrocitosis, su mecanismo de producción y por lo tanto los mecanismos que regulan la homeostasis del oxígeno, la clasificación de las eritrocitosis y finalmente analizar brevemente el grupo de eritrocitosis cuya etiología es la presencia de una hemoglobina anormal con alta afinidad por el oxígeno.
Fisiopatología de la eritropoyesis
1.- Eritropoyetina. Formación de eritrocitos
Se conocía desde la conquista de América por Hernán Cortés y Pizarro que el mal de altura se producía al transitar y pecnoctar los soldados españoles por estas elevadas altitudes (4). Este hecho se confirma en 1875 por D. Jourdanet, que demuestra como los sujetos que viven en lugares de elevada altitud, con bajas presiones de O2, tienen aumento de hematíes y la sangre más viscosa que los que habitan al nivel del mar. En 1906 P. Carnot y C de Flandre son los primeros que sugieren que la hipoxia genera un factor humoral que es capaz de producir eritrocitos a la que denominan “hemopoyetina”. AJ Erslev en 1953 describe esta hormona y la denomina eritropoyetina (EPO) y T Miyata en 1977 purifica la EPO en la orina de los pacientes anémicos. A lo largo de los años los avances en el campo de la biología molecular confirman que la hipoxia (niveles de oxígeno disminuidos, como sucede en la altitud) regula la producción de eritrocitos debido al incremento de EPO y establecen el mecanismo molecular de la vía de los sensores del oxígeno (5). El gen de la EPO se clonó en 1985.
La EPO se produce en las células intersticiales corticales peritubulares y su producción está en relación con el nivel de saturación de oxígeno de la hemoglobina. El mayor estímulo fisiológico para la producción de EPO es la disminución de aporte de oxígeno a los tejidos. Ante situaciones de hipoxia su secreción se incrementa centenares de veces con el consiguiente incremento de la eritropoyesis.
La EPO se une a su receptor (EPOR) en la superficie de los precursores eritroides (BFU-E), proeritroblastos y eritroblastos basófilos. La interacción de EPO con su receptor produce cambios conformacionales en EPOR, como su dimerización y su posterior fosforilización y asociación con la proteína citoplasmática tirosinkinasa “Janus Tyrosine kinase 2” (JAK 2) (6) JAK 2 se fosforiliza e inicia la señalización transcripcional de la vía STAT5, que así mismo se fosforiliza se transfiere al núcleo y activa numerosas vías de señalización y transcripción de señales, como la división y diferenciación de las células eritroides, y por otra parte a través de la activación de genes antiapoptóticos, como Bcl-XL inhibe la apoptosis de eritoblastos, regulando de este modo la producción y control de la masa eritrocitaria. La señalización de EPOR puede bloquearse por diferentes proteínas como SOCS o SHP-1 (4, 7, 8).
La expansión de las células precursoras eritroides se produce por un incremento de la activación transcripcional del gen EPO a nivel del elemento respondedor a hipoxia “hipoxia-responsive element “(HRE) localizado en la región 3’ promotora del gen EPO (9).
2.- Homeostasis del oxígeno. Vía de los sensores de oxígeno.
Las investigaciones realizadas por William G Kaelin, Peter J Ratcliffe y Gregg L Semenza, acerca de cómo las células detectan y se adaptan a los cambios de oxígeno les ha merecido en el año 2019 el Premio Nobel de Fisiología y Medicina.
La función fundamental de la hemoglobina es el transporte de O2 a los tejidos. Esta función la realizan merced a la capacidad de captar oxígeno a nivel de los alveolos pulmonares y liberarlo en los capilares de los diferentes tejidos.
El oxígeno es indispensable para el desarrollo y supervivencia de los seres vivos. Participa en la fosforilización oxidativa que transfiere la energía química almacenada en los enlaces de carbono de las moléculas orgánicas, al enlace de fosfato de alta energía del ATP, que se utiliza en las reacciones fisicoquímicas de las células (10). Los organismos han desarrollado estrategias para adaptarse a diferentes niveles de O2, hecho que se ha asociado con el desarrollo del sistema respiratorio, circulatorio o nervioso, diseñados para obtener y distribuir eficientemente el O2 a millones de células en el organismo de los seres superiores (10).
Aunque se sabía que la EPO era el mayor estímulo que regulaba la síntesis de la eritropoyesis, es gracias a los trabajos de Semenza, Kaelin y Ratcliffe como se identifica y lleva a cabo esta regulación a nivel celular (10, 11). La respuesta celular a la hipoxia está controlada por una serie de factores de transcripción denominados factores inductibles por hipoxia (HIF), que juegan un papel esencial en la homeostasis del oxígeno tanto durante el período embrionario, como en la vida postnatal. HIF está compuesto por dos unidades formando un heterodímero, HIFα y HIFβ, pertenecientes a la familia de PAS. Hay 3 isoformas de HIFα: HIF1α, HIF2α y HIF3α, que se sintetizan continuamente en respuesta a la hipoxia (12) HIF2α es la isoforma esencial en la regulación de EPO y juega un papel importante no sólo en la eritropoyesis, sino en la vascularización y en el desarrollo pulmonar (10). Tanto la isoforma HIF1α, como HIF2α contienen 2 dominios de transactivación a nivel N terminal (NTAD) y C terminal (CTAD). (11). HIF1 alfa se expresa en numerosas células, mientras que HIF 2 alfa se encuentra limitado a células endoteliales, cardiomiocitos, hepatocitos, células de la glia y células intersticiales del riñón.
En situaciones con tensión normal de oxígeno o normoxia una prolil hidrolasa 2 (PHD2) (13, 14) que utiliza O2 como sustrato, hidroliza los dos residuos de prolina de HIF2α en su región N terminal. Hay 3 isoenzimas PHD 1, 2 y 3 (12), pero PHD 2 es la que cataliza la hidrolización prolínica de HIFα. Los residuos hidrolizados de HIFa se unen a la proteína supresora de tumores von Hippel-Lindau (VHL) que es sensible a la ubiquitinación por el complejo E2-ubiquitin ligada y posteriormente degradado por el proteasoma celular (11), impidiendo por lo tanto su transcripción génica y la síntesis de EPO (12).
En situaciones de hipoxia, con cantidades limitadas de oxígeno, la hidroxilación de las subunidades de HIFα es muy lenta, escapa a la unión con la proteína VHL y por lo tanto a la degradación por el proteasoma. Se estabiliza y se acumula en el citoplasma, posteriormente se transfiere al núcleo en donde se dimeriza con HIFβ formando un heterodímero activo (15), que permite la expresión del gen EPO a nivel del DNA de la región del elemento respondedor a la hipoxia (HRE).
Por lo tanto la respuesta celular a la hipoxia está controlada por una familia de factores transcripcionales, factores inductibles por hipoxia o HIF, cuyo factor HIFα puede considerarse como el factor clave en la regulación de la homeostasis del oxígeno. La importancia de las tres proteínas HIF2α, PHD2 y VHL está sustentada por las eritrocitosis congénitas que se producen cuando existen mutaciones de estos tres factores.
HIFα no sólo actúa a nivel de la eritropoyesis, sino que además más de 300 genes se regulan transcripcionalmente por HIFα y están involucrados en la proliferación y supervivencia de células, angiogénesis, condrogénesis, adipogénesis, regulación del metabolismo de la glucosa, regulación del ciclo celular y además participa activamente en la regulación del hierro como síntesis de DMT1 (transportador de metales bivalentes 1), citocromo reductasa duodenal (Dcytb), transferrina, receptor de la transferrina (TFR1), ferroportina, síntesis del hemo (ALAS2) y producción de cadenas de globina (GATA-1) (10, 16).
Mención aparte merece la relación de HIF1α y HIF2α con la proliferación celular de las células cancerosas. Se han encontrado niveles aumentados de ambas proteínas en las biopsias tumorales de cánceres de vesícula, cerebro, mama, colón, cérvix, endometrio, cabeza y cuello, pulmones, ovario, páncreas, próstata, recto, y estómago (10). Semenza también ha demostrado que el incremento de la expresión de HIF1α aumenta el crecimiento tumoral, mientras que la pérdida de su actividad disminuye su crecimiento (17). En el momento actual drogas que inhiben HIF1 están siendo investigadas como dianas terapéuticas para el tratamiento de diferentes tumores sólidos (10).
3.- Transporte de oxígeno a los tejidos.
La oxigenación de los tejidos se realiza a través del oxígeno contenido en la hemoglobina, que se une al hierro en el estado ferroso en el bolsillo del hemo (1), próximo a la histidina distal, del segmento E de la cadena de globina (E7). Esta histidina impide que el oxígeno pueda penetrar al interior del hemo, unirse al hierro del hemo vecino y formarse metahemoglobina.
La hemoglobina está formada por un tetrámero (3), en donde la unión de la primera molécula de oxigeno facilita la entrada de las otras moléculas al resto de las cadenas de globinas. La primera molécula desestabiliza la configuración de la desoxihemoglobina, creando puentes de unión que facilitan la entrada de la siguiente molécula, y así de la tercera y cuarta. Esto constituye el efecto denominado hemo-hemo. Y se produce porque la hemoglobina puede tener dos configuraciones: deoxihemoglobina, forma tensa o T y oxihemoglobina forma relajada o R. La primera presenta baja afinidad por el oxígeno, mientras que la configuración R tiene una alta afinidad. Durante la oxigenación se produce un cambio alostérico de la forma T a la R, que permite un aumento considerable de la afinidad por el O2. En los pulmones la presión parcial de oxígeno es elevada y la Hb se satura al 100%, mientras que en los tejidos la presión parcial disminuye y el proceso de desoxigenación se produce de manera inversa, acelerándose la liberación de oxígeno a medida que se sueltan las moléculas de gas. Estos cambios configuracionales y reversibles de la captación y suelta de oxígeno pueden expresarse por la forma sigmoidea de la curva de disociación de la Hb, en la que la P50 representa la presión parcial de oxígeno en la cual el 50% de la Hb se encuentra saturada. La hemoglobina tetramérica del adulto tiene una P50 de 26 mm Hg (±1.6). Varios factores pueden influenciar la misma como CO2, temperatura, PH y el 2,3 bifosfoglicerato (2,3 BPG), factor que influye en gran medida en el desplazamiento de la curva de disociación de la Hb. Cuando se elimina el oxígeno se facilita la entrada del metabolito 2,3 BPG al interior del tetrámero de Hb (en la configuración desoxihemoglobina) Su aumento, al unirse con las dos cadenas de beta globina, desplaza la curva a la derecha, liberando el O2. Su disminución conlleva un aumento de la afinidad de la Hb por el oxígeno con desplazamiento hacia la izquierda (18).
Clasificación de las eritrocitosis
Las eritrocitosis pueden clasificarse en congénitas y adquiridas, y dentro de estas en primarias, es decir, con defectos intrínsecos de los hematíes y EPO independientes o secundarias debidas al exceso de la producción de eritropoyetina (Tablas 1,2, 3, y 4).
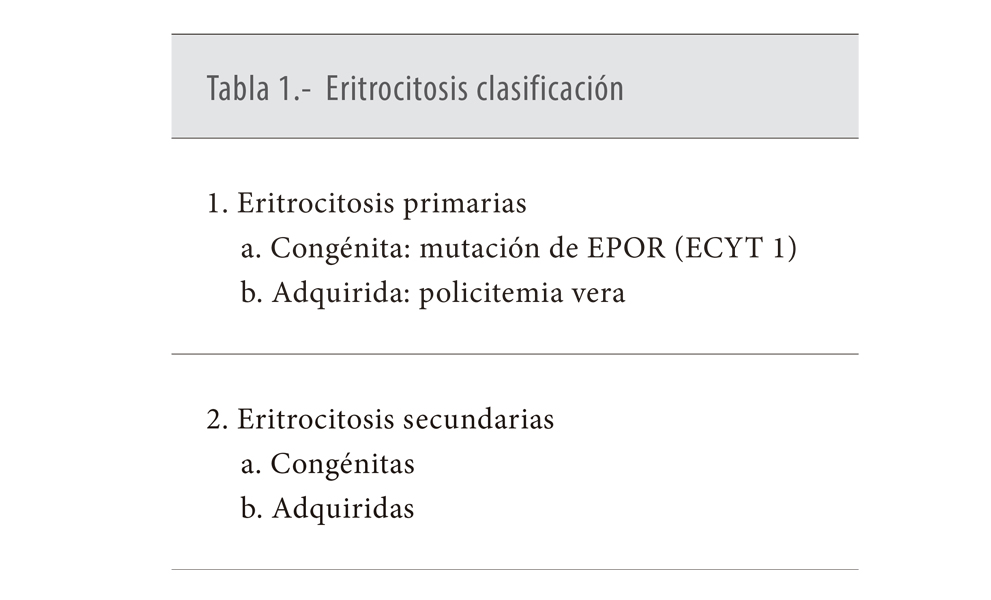



Dentro del primer grupo, eritrocitosis primarias congénitas, con un defecto intrínseco de los precursores eritroides, sólo se ha descrito la eritrocitosis congénita tipo 1 (ECYT 1), producida por la mutación del gen receptor de EPO (EPOR). Consiste en una eritrocitosis con bajos niveles de EPO, formación de colonias eritroides espontáneas sin la adicción de EPO al medio de cultivo e hipersensiblidad de los progenitores hematopoyéticos a la eritropoyetina (19). Los pacientes son generalmente asintomáticos o con síntomas leves, sin embargo se han descrito pacientes con complicaciones severas como hipertensión arterial, trombosis venosas profundas y enfermedad coronaria (Tabla1).
La enfermedad encuadrada dentro de las eritrocitosis primarias adquiridas, la constituye la policitemia vera (PV) que se engloba dentro de los síndromes mieloproliferativos crónicos Philadelphia negativos (2, 3, 20). En 95% de los casos de PV se ha encontrado una mutación somática del gen JAK2, en el exón 14 que genera el cambio de valina por fenilalanina en el aminoácido 617 de la cadena proteica. En un 3% de los casos la mutación se produce en el exón 12 (20). Los criterios diagnósticos de la PV se han modificado en el año 2016 por la OMS, con el objeto de establecer el diagnóstico precoz con la trombocitosis esencial, que en un 50% de los casos puede tener mutación del JAK2 y que en los estadios iniciales también puede cursar con leve eritrocitosis. Las cifras de hemoglobina son diagnósticas con >16.5 g/dl en el varón, >16 g/dl en la mujer y el valor hematocrito >49% en varones y 48% en mujeres, si además se cumplen los demás criterios (Tabla 4).
Las eritrocitosis secundarias pueden ser congénitas y adquiridas. En ellas no existen defectos intrínsecos de los hematíes y tienen niveles de eritropoyetina normales o elevados.
Dentro de las congénitas (ECYT) en primer lugar contemplamos un grupo de eritrocitosis en las que se describen mutaciones de genes en relación con la homeostasis del oxígeno (21) (Tabla 2):
- Eritrocitosis congénita tipo 2 (ECYT 2), es la producida por la mutación del gen supresor de tumor Von Hippel-Lindau (gen VHL). Los primeros casos fueron descritos en la región rusa de Chuvash, en donde es endémica (7, 22, 23). Se han descrito casos en otros grupos étnicos, con diferentes mutaciones del gen VHL. En la región de Chuvash se produce por el cambio de arginina por triptófano en el aminoácido 598 de la proteína VHL, en estado homocigoto. En la sintomatología puede observarse rubor, hemangiomas vertebrales y eventos trombóticos arteriales y venosos (19).
- Eritrocitosis congénita tipo 3 (ECYT 3). Está producida por la mutación del gen EGLN1, que codifica la proteína PHD2 (19, 23). Se han descrito 24 casos. La herencia es autosómica dominante (19).
- Eritrocitosis congénita tipo 4 (ECYT 4). Se trata de una mutación del gen EPAS1, que codifica el factor de transcripción, factor inductible por hipoxia, HIF2α. La mutación consiste en un cambio de aminoácido (G537W) producida por la mutación del gen en el exón 12 (1609 G>T). Es autosómica dominante. Se han descrito otras mutaciones con diferentes variantes de HIF2α (24). El cambio de aminoácido impide la hidrolización de los residuos de prolina de HIF2α y por lo tanto su unión a la proteína VHL y la destrucción por el proteasoma. Los niveles de EPO están elevados.
Algunos pacientes de este grupo de enfermedades congénitas se acompañan de tumores neuroendocrinos (15).
Hemoglobinas con alta afinidad por el oxígeno
La primera descripción de una hemoglobinopatía con alta afinidad por el oxígeno la realizó Charache en el año 1966 (25) en un paciente de 81 años con Hb de 19.9 g/dl, y la denominó Hb Chesapeake. Desde entonces se han descrito más de 100 variantes con un aumento de la afinidad de la Hb por el oxígeno, que comentaremos posteriormente más ampliamente.
Una eritrocitosis congénita extraordinariamente rara, sólo se han descrito 3 casos en la literatura (23), se debe a la mutación del gen codificante de la enzima 2,3 difosforigleromutasa, que actúa en la vía glicolítica a través del ciclo de Rappaport-Luebering y cataliza la conversión de 1,3 BPG en 2,3 BPG, y por lo tanto al disminuir el 2,3 BPG aumenta la afinidad de la Hb por el oxígeno, con poca cesión a los tejidos, hipoxia e incremento de EPO y eritrocitosis.
Finalmente la metahemoglobina también puede producir cianosis y eritrocitosis.
Eritrocitosis secundarias adquiridas
Son las más frecuentes y afectan a un gran número de pacientes en la clínica médica. Se caracterizan por tener una EPO normal o aumentada, y de ahí que también se las denomine EPO dependientes. Los precursores son normales, así como las vías congénitas de regulación de EPO y de los sensores de oxígeno (Tablas 2,3 y 4).
En general son consecuencia de una producción de EPO aumentada. Las más frecuentes se originan como consecuencia de hipoxia y presentan una respuesta apropiada a la síntesis de EPO. Otras son consecuencia de la producción ectópica de EPO, sin hipoxia, y son debidas a la síntesis de EPO neoplásica (1, 4).
Diagnóstico diferencial
Ante una eritrocitosis confirmada lo primero que hay que excluir es la policitemia vera (PV), dado que es una enfermedad clonal que en un 30% y 10% de los casos respectivamente puede evolucionar a mielofibrosis o a leucemia aguda mieloblástica, con una supervivencia disminuida en relación con la población normal (2, 20).
Los criterios para el diagnóstico de PV, en el momento actual, son los dictados por la OMS en el año 2016 (Tabla 5). Hay tres criterios mayores y un criterio menor. Para el diagnóstico se requiere la presencia de los 3 criterios mayores o de 2 criterios mayores junto al criterio menor (20).
Por lo tanto, ante una eritrocitosis absoluta, además de una historia clínica detallada reseñando antecedentes familiares y una exploración física completa, hay que confirmar en el hemograma nuevamente la existencia de eritrocitosis y valorar si existen otras citosis. En los datos de bioquímica no hay que olvidar ácido úrico, función renal, LDH, perfil hepático y sedimento urinario; así como determinación de EPO y de JAK2, y la gasometría arterial (saturación de O2).
Si la EPO es baja y existen otros criterios de PV como JAK positivo, etc., contemplar el diagnóstico de PV.
Si hay historia familiar y la EPO es normal o elevada, por ser más frecuentes que el resto de las eritrocitosis congénitas, descartar hemoglobinopatías con alta afinidad por el oxígeno y por lo tanto, realizar P50 y secuenciación de cadenas de globina. Pensar también en la vía de los sensores de O2 (1, 4, 19).
Dada la frecuencia de las eritrocitosis adquiridas secundarias en relación con la hipoxia, realizar una saturación arterial de O2, y si es menor del 92-94%, pensar en enfermedad pulmonar. Deben realizarse estudios radiológicos, radiografía de tórax, ecografía abdominal (en este caso para descartar enfermedades renales como poliquistosis, hidronefrosis, tumores y trasplante renal). Son causa frecuente de eritrocitosis las enfermedades pulmonares obstructivas y el síndrome de hipoventilación pulmonar, así como las comunicaciones cardiacas derecha izquierda o bien los “ shunt” pulmonares.
Así mismo, realizar carboxihemoglobina por la frecuencia de eritrocitosis asociada al hábito tabaquismo y metahemoglobina y polisomnografía por ser la apnea del sueño (15) una causa común de eritrocitosis. La polisomnografía registra la saturación de oxígeno durante el sueño y puede demostrar saturación de O2 normal diurna, y sin embargo una reducción del 20% durante el sueño (1). Esta asociación de hipoxemia nocturna y eritrocitosis no se ha confirmado en algunos estudios (26).
En el síndrome de TEMPI se ha demostrado eritrocitosis con EPO elevada, telangectasias, gammapatía monoclonal, y “shunt” intrapulmonar (1, 4). La etiología no es bien conocida, pero el hecho que pueden mejorar con bortezomib puede deberse al efecto de la paraproteína sobre algún factor regulador de HIF (27).
Numerosos tumores benignos y malignos pueden producir EPO ectópica, y con la consiguiente eritrocitosis. Dentro de los benignos miomas uterinos, hemangioblastomas del sistema nervioso central, y malignos como hepatocarcinoma, carcinoma renal….
Por lo tanto, el diagnóstico diferencial de las eritrocitosis debe de hacerse en una bien documentada historia familiar y personal, con la realización de exploraciones pertinentes (ecografía, resonancia magnética, TAC y polisomnografía…), y determinaciones como saturación de oxígeno, EPO, estudio de JAK2, P50 y estudios de biología molecular (28)(Tabla 5).
Aun así, persiste un grupo de eritrocitos idiopáticas que no se conoce la causa y por lo tanto tampoco el tratamiento. Afortunadamente con una buena planificación y el estudio de paneles de secuenciación masiva cada vez son menores. Algunos autores incluso dudan de su existencia (3).
Hemoglobinopatías por alta afinidad de la hemoglobina por el oxígeno
Las hemoglobinopatías son los trastornos monogénicos más comunes en la población mundial y pueden ser debidos a defectos de síntesis de una o más cadenas de globina –talasemias- o bien a la síntesis anormal de una cadena, generalmente por cambio de un aminoácido por otro y son las hemoglobinopatías estructurales o variantes de hemoglobina. Se han descrito más de 1000 variantes de hemoglobina normal. El 95% de ellas se deben a la sustitución de un aminoácido en las cadenas α o β de globina y en menor número en las γ. Dependiendo de la naturaleza del aminoácido mutado y de su posición dentro de la cadena de globina pueden producirse alteraciones en la solubilidad, la estabilidad o bien en la función de la hemoglobina. Sin embargo más de la mitad son asintomáticos, tanto clínica como fenotípicamente.
Cuando los cambios se producen en aminoácidos que involucran a la estructura de la hemoglobina, que son críticos para la función transportadora de oxígeno, se puede producir alteración de la afinidad de la Hb por el O2, bien por exceso (18,29) o bien por baja afinidad, en ocasiones con cianosis (30).
En las hemoglobinopatías con alta afinidad por el oxígeno hay falta de liberación del mismo en los tejidos y por lo tanto hipoxia tisular, con incremento en la síntesis de EPO y consecuentemente eritrocitosis secundaria.
En la estructura cuaternaria de la hemoglobina existen aminoácidos muy estables y críticos, cuyo cambio puede desestabilizar la configuración desoxi-oxihemoglobina y por lo tanto la curva de disociación de la Hb y la P50. Cambios de aminoácidos a nivel de la unión α1β2 (α1, β1) en la región C terminal, en puntos de unión de 2,3 BPG o en aminoácidos ubicados en el bolsillo del hemo pueden producir aumento de la afinidad de la Hb por el O2, con P50 disminuida (18, 31).
Se han descrito alrededor de 100 mutaciones diferentes que producen incremento de la afinidad, la mayoría en la cadena β. Sólo un 20% afectan a la cadena α.
En el estudio es fundamental valorar la historia familiar, dado que pueden observarse varios miembros de la familia afectos, con eritrocitosis. La herencia es autosómica dominante.
Para su tipificación es necesario realizar estudio convencional de hemoglobinas, HPLC de intercambio iónico, HPLC de cadenas de globina y secuenciación de la cadena de globina mutada. Alrededor de un 30% de los casos tienen el estudio de hemoglobinas alterado, mientras que siempre se separa la cadena de globina mutada por HPLC de fase reversa.
Es obligatorio realizar la P50, que en todos los casos está disminuida, y el estudio molecular de la mutación (18).
En el Servicio de Hematología del Hospital Clínico San Carlos de Madrid, hemos estudiado 34 pacientes correspondientes a 17 familias españolas no relacionadas entre sí con 10 diferentes hemoglobinopatías con alta afinidad por el oxígeno. Estos pacientes han podido controlarse durante más de 10 años, y ninguno ha presentado complicaciones trombóticas venosas o arteriales. Tampoco se ha observado, en pacientes embarazadas, pérdidas fetales.
De los 10 tipos diferentes de hemoglobinopatía, 8 ya habían sido descritas previamente y dos (Hb Badalona y Hb La Coruña) (32, 33), las hemos caracterizado molecularmente por primera vez en el laboratorio del Clínico San Carlos.
Cuatro familias (6 casos) presentan Hb San Diego, cinco familias (13 casos) tienen Hb Johnstown, las restantes hemoglobinopatías sólo se han descrito en una familia.
En cuatro de ellas el cambio del aminoácido se produce en la zona de unión α1β2, en donde se realiza el cambio de configuración de la molécula durante la oxigenación, de T a R (Hb San Diego, Hb Johnstown. Hb Malmô, Hb Columbia-Missuri). En dos variantes Hb Strasbourg y Hb Siracuse, por cambio de un aminoácido en la unión con el 2,3 BPG. En las hemoglobinopatías de origen español –Hb Badalona y Hb La Coruña- en la región de contacto con el hemo, y en la Hb Bethesda la mutación afecta a la zona de contacto α1β1.
La hemoglobina Oympia con cambio de valina por metionina en el aminoácido 20 de la cadena β, también produce eritrocitosis. Aunque el aminoácido 20 de la cadena β está situado en la superficie externa de la molécula, y no parece afectar a ninguna región crítica del transporte de O2, el cambio de un aminoácido polar, valina, por uno hidrofóbico como metionina, puede indirectamente alterar alguna unión crítica en esta zona (18). Otra hemoglobinopatía Hb Trollhättan, que también cambia el aminoácido 20 (Val→Glu) presenta así mismo aumento de la afinidad de la Hb por el O2 y eritrocitosis.
En dos familias la hemoglobinopatía estructural se asocia con talasemia. En la primera la hemoglobina anormal Johnstown con βº talasemia (Intron 1 nt1). Los pacientes presentan Hb de 18-19 g/dl con VH del 60% y una P50 de 13-15 mm Hg. No se observa Hb A normal y prácticamente la totalidad de Hb es la variante Johnstown; sin embargo están asintomáticos.
En la segunda familia (familia nº 18) el recién nacido es portador de Hb Andrew-Mineápolis, junto con δβº talasemia (34). Se detectó en un programa de escreening neonatal al observar ausencia de Hb A normal. A los 10 días del nacimiento los valores de la variante eran de 55%, con Hb fetal 43% y P50 17 mmHg. El padre es portador de δβº talasemia Spanish y la madre es totalmente normal. Es por lo tanto una mutación de Hb Andrew-Mineápolis “de novo”. La paternidad se demuestra con el polimorfismo PvuII (3’HVR, sonda α)El estudio posterior realizado a los 42 meses demuestra eritrocitosis (VH 55%, Hb 17,8 g/dl) con Hb anormal de 40% y Hb F 57,5% y P50 17 mmHg. En la Hbpatía Andrew-Mineápolis [β144 (HC1) Lys-Asn] hay una sustitución en el aminoácido 144 de la cadena β de lisina por asparragina con lo que se alteran los puntos de unión de hidrogeno con el imidazol de la histidina C terminal (34).
Es muy infrecuente que la eritrocitosis se detecte en niños, salvo en casos de estudio familiar o como en nuestro caso, cuando están asociadas a talasemia.
De las 11 mutaciones que describimos, 10 pertenecen a la cadena β y solamente una afecta a cadena α (Hb Columbia-Missuri).
Los pacientes con hemoglobinopatías con alta afinidad por el oxígeno no suelen manifestar importantes manifestaciones clínicas. Se han descrito leves cefaleas, mareos en relación con la hiperviscosidad, pero nunca muy relevantes y la mayoría de las veces no requieren tratamiento médico.
CONCLUSIONES
En este trabajo, hemos revisado las diferentes causas de eritrocitosis, así como su diagnóstico diferencial y las técnicas o estudios necesarios para su catalogación. Se ha realizado una puesta a punto de los mecanismos fisiopatológicos que controlan la eritropoyesis, haciendo un particular análisis de los mecanismos que regulan la homeostasis del oxígeno y la vía de los sensores de oxígeno.
Dado que el objetivo fundamental del diagnóstico diferencial de las eritrocitosis es realizar el correcto diagnóstico de la policitemia vera, señalamos los criterios diagnósticos propuestos por la OMS en el año 2016.
Finalmente aportamos nuestra experiencia en las hemoglobinopatías con alta afinidad por el oxígeno y sus mecanismos de producción.
BIBLIOGRAFÍA
- Arrizabalaga Amuchastegui B. Eritrocitosis secundarias adquiridas. Eritrocitosis idiopática. En “Eritropatología”. Coordinadores: Beatriz Arrizabalaga, F. Ataulfo González, Ángel Remacha. Barcelona. Ambos Mark. 2017; pág 501-513.
- Spivak JL. Myeloproliferative neoplasms. N Engl J Med 2017; 376(22): 2168-2181.
- Patnaik MM, Tefferi A. The complete evaluation of erythrocytosis: congenital and acquired. Leukemia 2009; 23: 834-844.
- Guillermo Martín Núñez. Eritrocitosis, diagnóstico diferencial y tratamiento. LXI Congreso Nacional SEHH. Congreso Nacional SETH. Programa educacional 2019; pág: 7-15.
- Wang GL, Semenza GL. Molecular basis of hipoxia-induced erythropoietin expresión. Curr Op. Hematol 1996; 3(2): 156-162.
- Kuhrt D, Wojchowski DM. Emerging EPO and EPO receptor regulators and signal transducers. Blood 2015; 125: 3536-3541.
- Mc Mullin MF. Congenital erythrocytoses. Int J Lab Hematol 2016; 38(1): 59-65.
- Maia TM, Bento C, Ribeiro LM. Eritropoyesis en “Eritropatología”. Coordinadores: Beatriz Arrizabalaga, F. Ataulfo González, Ángel Remacha. Barcelona. Ambos Mark 2017; pág 3-16.
- Koulnis M, Porpiglia E, Hidalgo D, Socolovsky M. Erythropoieis from molecular pathways to system properties. Adv Exp Med Biol 2014; 844: 37-58.
- Semenza GL. Hypoxia-inducible factors in Physiology and Medicine. Cell 2012; 148(3): 399-408. Doi 10.1016/j cds. 2012.01.021
- Kaelin WG, Ratcliffe PJ. Oxygen sensing by metazoans the central role of the HIF dydroxylase pathway. Mol Cell 2008; 30: 393-402.
- Jiang BH, Rue E, Wang GL, Rose R, Semenza GL. Dimerization, DNA binding and transactivation propieties of hipoxia-inductible factor 1. J Biol Chem 1996; 271: 17771-17778.
- Kaelin WG. Proline hydroxylation and gene expresión. Annu, Rev Biochem 2005; 74: 115-118.
- Percy MJ, Zhan Q, Flores A et al. A family with erythrocytosis establishes a role for prolyl hydroxglase domain protein 2 in oxygen homeostasis. Proc Natl Acad Sci USA 2006; 103: 654-659.
- Franke K, Gassmann M, Wieloclx B. Erythrocytosis: The HIF patway in control. Blood 2013; 122(7): 1122-1128.
- Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inductible nuclear factors bind to an enhancer element locate 3’ to the human erythropoietin gene. Proc Natl Acad Sci USA 1991; 88: 5680-5684.
- Semenza GL. Defining the role of hipoxia-inductible factor 1 in cáncer biology and therapeutics. Oncogene 2010; 29: 625-634.
- González Fernández FA, Villegas A, Ropero P et al. Haemoglobinopathies with high oxygen affinity. Esperience of Erythropathology Cooperative Spanish Group. Ann Hematol 2009; 88: 325-328.
- Bento C, Percy MJ, Gardie B et al: On behalf of ECE-Consortium. Genetic basis of congenital erythrocytosis: mutation update on line databases Hum Mutat 2014; 35: 15-26.
- Barbui T, Tefferi A, Vannuchi AM et al. Philadelphia chromosome-negative classical myeloproliferative neoplasms: revised management recommendations fron European Leukemia Net. Leukemia 2018; 32(3): 1057-1069.
- Lee FS, Percy MJ. The HIF pathway and erythrocytosis. Annu Rev Pathol 2011; 6: 165-192.
- Cario H, Schwartz R, Jorch N. Mutations in the von Hippel-Lindau (VHL) tumor suppressor gene and VHL-haplotype analysis in patients with presumable congenital erythrocytosis. Haematologica 2005; 90(1): 19-24.
- Cario H, McMullin F, Bento C et al. Erythrocytosis in children and adolescents: classification, characterization and consensus recommendations for the diagnostic approach. Pediatr. Blood Cáncer 2013; 60(11): 1734-1738.
- Percy MJ, Furlow PW, Lucas GS et al. A gain of function mutation in the HIF2α gene in familial erythrocytosis. N Engl J Med 2008; 358(2): 162-168.
- Charache S, Weatherall DJ, Clegg JB. Policythemia associated with a hemoglobinopathy. J Clin Invest 1966; 45: 813-822.
- Solmac S, Kuksal F, Ganidagli S. Is obstructive sleep apnea syndrome really one of the causes of secundary polycythaemia. Hematology 2015; 20(2): 108-111.
- Sykes DB, Schroyens W. Complete responses in the TEMPI Syndrome after treatment with Daratumumab. N Engl J Med 2018; 378(23): 2240-2242.
- Lee G, Arcasoy MO. The clinical and laboratory evaluation of the patient with erythrocytosis. J Int Med 2015; 26: 297-302.
- Torres W, García Roa M, Manubens A et al. Hemoglobinopathies with high oxygen affinity. Haematologica 2015; 99(51): 183.
- Torres W, García Roa M, Gutiérrez Alvariño M et al. Characterization of 6 hemoglobinophaties ocurring with cianosis and/or lowering the oxygen saturation. Haematologica 2015; 99(51): 183.
- Wajcman H, Galacteros F. Hemoglobin with high oxygen affinity leading to erythrocytosis. New variants and new concepts. Hemoglobin 2005; 29(2): 91-106.
- Juncá J, Villegas A, Ropero P, Polo M, Valverde F. Characterization of a new hemoglobin variant: Hb Badalona β(31) Leu→val). Ann Hematol 2002; 81(4): 179-181.
- Ropero P, Fernández Lago C, Villegas A et al. Hb La Coruña [β38 (C4) Thr→Ille] a new hemoglobin leading to familial polycythemia. Hemoglobin 2006; 30(3): 379-383.
- Ropero P, González FA, Cela E et al. Erythrocytosis in a child due to Hb Andrew-Minneapolis [β 144 (HC1) Lys→ASN (AAG>AAT o AAC] associated with a Spanish (δβ)º thalassemia. Hemoglobin 2013; 37(1): 48-55.
DECLARACIÓN DE TRANSPARENCIA
Los autores/as de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en la presente revisión.
ranm tv
Ana Villegas Martínez
Real Academia Nacional de Medicina de España
C/ Arrieta, 12 · 28013 Madrid
Tlf.: +34 91 159 47 34 | E-Mail: anamaria.villegas@salud.madrid.org
Año 2020 · número 137 (01) · páginas 35 a 43
Enviado*: 03.03.20
Revisado: 10.03.20
Aceptado: 24.03.20
* Fecha de lectura en la RANM