

Resumen
La enfermedad de células falciformes (ECF) es una enfermedad monogénica con un fenotipo altamente variable que, depende esencialmente de la cantidad de hemoglobina fetal (HbF), constituyendo ésta el principal modulador. La variación de los niveles de HbF entre los pacientes está regulada genéticamente.
La HbF determina tanto el fenotipo de la enfermedad como la respuesta al tratamiento con el principal fármaco utilizado, la hidroxiurea (HU). Los esfuerzos de los investigadores se han centrado en descubrir los factores genéticos responsables de su variación; describiéndose principalmente los haplotipos del clúster β y SNPs (del inglés Single Nucleotide Polymorphism, SNPs) en tres loci diferentes: BCL11A, HBS1L-MYB y el clúster β-globina.
Objetivo: Determinar en una cohorte de pacientes con ECF, la posible relación entre el número de SNPs y los haplotipos con mayores niveles de HbF. Una asociación positiva podría explicar porqué ciertos haplotipos, como el Senegal o el Árabe-Indio, tienen mayores niveles de HbF y una enfermedad menos grave.
Métodos: Para comprobar esta hipótesis se llevó a cabo la caracterización de los haplotipos mediante la técnica PCR-RFLP y el genotipado de tres polimorfismos puntuales de un solo nucleótido (del inglés Single Nucleotide Polymorphism, SNPs) representativos de los tres loci con mayor asociación con la variación de HbF: XmnI (rs7482144), BCL11A (rs4671393) y HBS1L-MYB (rs9376092).
Resultados: Encontramos un mayor número de SNPs en los haplotipos relacionados con mayor HbF y menor número en los que presentan menos HbF, aunque sólo el SNP XmnI (rs7482144) ha demostrado asociación estadísticamente significativa.
Conclusiones: Encontramos relación directa entre los haplotipos y el número de SNPs. Los haplotipos con niveles más elevados de HbF y fenotipo menos severo, presentaban mayor número de SNPs. De modo que, los haplotipos Benín y Bantú asociados tradicionalmente con mal pronóstico, son los que han mostrado un menor número de SNPs mutados.Abstract
Sickle cell disease (SCD), despite being a monogenic disease, presents a highly variable phenotype that essentially depends on the amount of fetal hemoglobin (HbF), constituting the main modulator of the disease. The variation of HbF levels between patients is genetically regulated.
HbF determines both the phenotype of the disease and the response to treatment with the main drug used, hydroxyurea. Researchers’ efforts have focused on discovering the genetic factors responsible for its variation; mainly describing the haplotypes of the β cluster and SNPs in three different loci: BCL11A, HBS1L-MYB and the β-globin cluster.
Objective: To determine, in a cohort of patients with SCD, the possible relationship between the number of SNPs and haplotypes with higher levels of HbF. A positive association could explain why certain haplotypes, such as Senegal or Arab-Indian, have higher levels. of HbF and less severe disease.
Methods: To verify this hypothesis, the characterization of the haplotypes was carried out using the PCR-RFLP technique and the genotyping of three SNPs representative of the three loci with the greatest association with the variation of HbF: XmnI (rs7482144), BCL11A (rs4671393) and HBS1L-MYB (rs9376092).
Results: A greater number of SNPs has been reported in the haplotypes related to higher HbF and a lower number in those with less HbF, although only the SNP XmnI (rs7482144) has shown a statistically significant association.
Conclusions: A direct relationship between haplotypes and the number of SNPs has been found, reporting that haplotypes with higher levels of HbF and thus a less severe phenotype, had a greater number of SNPs. Thus, the Benín and Bantu haplotypes traditionally associated with poor prognosis are the ones that have shown a lower number of mutated SNPs.Palabras clave: Enfermedad de células falciformes; Haplotipos del clúster β; XmnI; BCL11A; HBS1L-MYB; Hemoglobina Fetal.
Keywords: Sickle cell disease; Haplotypes of the β cluster; XmnI; BCL11A; HBS1L-MYB; Fetal Hemoglobin.
INTRODUCCIÓN
La enfermedad de células falciformes (ECF) es un trastorno hereditario con una fisiopatología compleja impulsada en gran medida por la vasooclusión y la anemia hemolítica. Los pacientes con ECF pueden experimentar gran variedad de síntomas y complicaciones, incluido el síndrome torácico agudo, infecciones, hipertensión pulmonar, accidente cerebrovascular y crisis vasooclusivas dolorosas (1).
La enfermedad es causada por una transversión de un solo nucleótido en el codón 6 GAG> GTG (HBB: c.20A>T) (NM_000518.4) del gen β globina (HBB), que conduce a la producción de la variante de hemoglobina más común en todo el mundo, la HbS, caracterizada por la sustitución del aminoácido Glu→Val en la posición β6(A3) (2) . Además, se han caracterizado, al menos, cinco haplotipos diferentes en el cluster β, lo que sugiere distintos orígenes geográficos del mismo gen de βS (Senegal, Benín, Bantú, Árabe-Indio y Camerún), documentando que los haplotipos diferían en los niveles de Hb Fetal (HbF) (3,4).
La ECF (OMIM 603903) es uno de los trastornos autosómicos recesivos más comunes en todo el mundo con más de 300.000 recién nacidos afectados cada año, esperando que sean más de 400.000 para el 2050 (5). En Europa se estima que la prevalencia de la ECF en los 27 estados miembros es de aproximadamente 1/150. Y en España en el Registro Sickle Cell Disease (SCD) [Enfermedad de Células Falciforme] hay inscritos 1.142 casos. La detección temprana, tratamientos profilácticos con penicilina y vacunas han mejorado la calidad y aumentado la esperanza de vida de pacientes con ECF, aunque el único tratamiento curativo es el trasplante alogénico de donante HLA compatible (6).
A pesar de que todos los pacientes homocigotos para el alelo HbS tienen el mismo genotipo (βS/βS), la gravedad de la enfermedad puede ser enormemente variable entre los sujetos afectados, observándose pacientes con una clínica severa, hasta casos en donde los síntomas son mucho más leves. La marcada heterogeneidad fenotípica se debe tanto a determinantes genéticos como ambientales, siendo los principales, entre otros, la presencia de HbF (3,7). La HbF es considerada como un modulador de las características clínicas y hematológicas de la ECF (1,2).
Sólo existe un fármaco aprobado por la FDA y la EMA que induce la producción de HbF, la hidroxiurea (HU), pero no todos los pacientes consiguen aumentar los niveles de HbF y mejorar. Aunque la mayoría de los pacientes tratados con este fármaco responden adecuadamente, existe entre un 10 y un 20% de adultos que muestran una respuesta mínima (8,9)
Esta variabilidad probablemente sea debida entre otras causas, a los niveles basales de HbF que varían entre los haplotipos del clúster β, a la heterogeneidad en los genes responsables del metabolismo de la HU y a los locus de rasgo cuantitativo (QTLs del inglés quantitative trait locus) que afectan a la expresión de los genes γ-globina (HBG) entre los que se encuentran XmnI del locus de β-globina en el cromosoma 11p15, BCL11A en el cromosoma 2p15 y la región intergénica HBS1L-MYB en el cromosoma 6q23. La variante XmnI (rs7482144) ejerce un efecto directo en la expresión del gen HBG2 (10), mientras que las variantes en BCL11A (rs4671393) y en la región intergénica HBS1L-MYB (rs9376092) aumentan los niveles de HbF al disminuir la expresión de BCL11A y MYB, respectivamente, represores transcripcionales de la síntesis de cadenas γ-globina (7).
El objetivo principal de este trabajo ha sido ver si existe una relación directa entre los SNPs XmnI (rs7482144) localizado en HBG2, rs4671393 localizado en BCL11A y rs9376092 localizado en HBS1L-MYB y los haplotipos y observar si los haplotipos relacionados con la menor gravedad, por tener valores aumentados de HbF, presentan un mayor número de estos SNPs. Para cumplir este objetivo, primero se tuvo que analizar la asociación estadística del porcentaje de Hb F para cada SNPs y después el número y frecuencia de los SNPs en cada haplotipo.
MATERIAL Y MÉTODOS
Se han estudiado un total de 28 pacientes diagnosticados de ECF (14 mujeres y 14 hombres), sin tratamiento con HU y mayores de 6 años (media de edad 11 años). Las muestras fueron recibidas en el Hospital Clínico San Carlos de Madrid durante los años 2019 y 2020 procedentes de diferentes regiones españolas.
A todos se les realizó un estudio hematimétrico con recuento de reticulocitos (Coulter LH750 Analyzer; Beckman Coulter, Brea, CA, EE. UU.) y morfología de los glóbulos rojos. Los niveles de HbA2 y HbF fueron medidos por cromatografía líquida de intercambio iónico de alta resolución (HPLC-CE) (VARIANTTM; Bio-Rad Laboratories, Hercules, CA, EE. UU). Las hemoglobinas fueron estudiadas por electroforesis capilar de zona (EC) [Sebia Capillarys Flext (Sebia, Norcross, GA)] y HPLC-CE utilizando el programa corto para la β-talasemia de BioRad (Bio-Rad, Hercules, CA) según instrucciones del fabricante.
Tras el aislamiento de ADN genómico (Biorobot Ò EZ1; Quiagen GmbH, Hilden, Alemania), el ADN fue cuantificado en un fluorímetro Invitrogen Qubit 4 (Thermo Scientific, Wilmington, DE, EE. UU). Se descartó la asociación con α talasemia mediante cribado de las mutaciones puntuales y deleciones de α-talasemia más comunes en el mundo (21 en total) mediante PCR multiplex seguida de hibridación inversa con el kit comercial Alpha-Globin StripAssay (ViennaLab Diagnostic GmbH, Viena, Austria) cuya sensibilidad clínica es > 90%.
La caracterización molecular de la HbS se realizó con el kit comercial β-Globin StripAssay MED (ViennaLab Diagnostic GmbH, Vienna, Austria) y su confirmación mediante secuenciación automática de Sanger del gen β-globina siguiendo el protocolo previamente descrito (11). Los haplotipos del clúster β se obtuvieron mediante amplificación y digestión con enzimas de restricción (PCR-RFLP) según el protocolo descrito por Rahimi et al. (12).
El genotipado de los SNPs localizados en el gen Gγ (rs7482144); en el gen BCL11A (rs4671393) y en la región intergénica HBS1L-MYB (rs9376092) se llevó a cabo por secuenciación automática de Sanger utilizando los primers que aparecen recogidos en la Tabla 1.

En el estudio descriptivo de los datos las variables cualitativas se presentan con su distribución de frecuencias. Las variables cuantitativas se resumen con su media y desviación estándar (DE). Las variables cuantitativas que muestran una distribución asimétrica se resumen con la mediana y el rango intercuartílico (RIQ). En la comparación de los parámetros entre los grupos de estudio se evalúa la asociación mediante el test no paramétrico de Fisher debido a que los grupos tienen un pequeño tamaño muestral. Para todas las pruebas se acepta un valor de significación del 5%. El procesamiento y análisis de los datos se realiza mediante el software estadístico IBM SPSS Statistics v.2º.
Todos los índices hematológicos y hallazgos clínicos fueron llevados a cabo con el consentimiento informado previo de los pacientes y el estudio fue aprobado por el Comité de Ética del Hospital Clínico San Carlos, Madrid, España. Todos los experimentos fueron realizados de acuerdo con la Declaración de Helsinki.
RESULTADO
El valor medio de los datos hematológicos queda recogido en la tabla 2. Respecto a los valores de obtenidos del estudio de las hemoglobinas fueron: HbA2 [2,62 ± 0,52%], HbF [14,95 ± 9,13%] y HbS [81,86 ± 8,46%].

Los haplotipos se han inferido en base a la presencia (+) o ausencia (-) de corte en los sitios polimórficos por enzimas de restricción específicas [5’ε-HincII; 5’Gγ-XmnI; GγIVSII-HicIII; AγIVSII-HicIII; 3’ψβ-HincII; β-AvaII]. En la población de estudio se han reportado los haplotipos africanos Benín (—-++), Bantú (–+–+), Senegal (-++-++) y Camerún (–++++). El haplotipo mayoritario de la muestra ha sido Benín (70%), seguido de Bantú (15%), Senegal (11%) y Camerún (4%).
Los haplotipos con los valores de HbF más altos son Benín: 16,59 ± 9,44%, y Senegal: 14,52 ± 4,76%, mientras que el haplotipo Bantú tiene los valores de HbF más reducidos: 5,94 ± 2,42 %.
La frecuencia de los SNPs estudiados aparece en la tabla 3. Siendo los más frecuentes el estado homocigoto wild type para XmnI (rs7482144) y HBS1L-MYB (rs9376092) con un 88.9% y un 77.8% respectivamente.

La variación de la HbF asociada a los tres SNPs, en los tres genotipos posibles (homocigoto para el alelo wild-type, homocigoto para el alelo mutado y heterocigoto) se ilustra en la Figura 1.
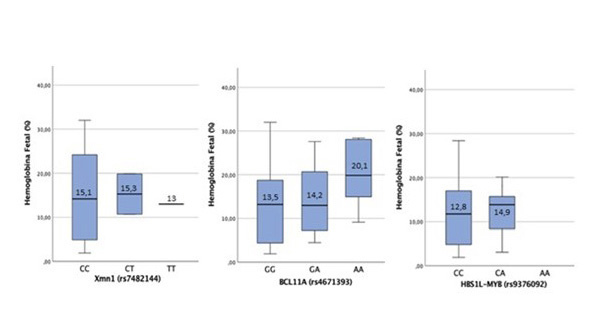
La distribución de HbF entre los genotipos del SNP XmnI (rs7482144) muestra que los valores de HbF por debajo del 10% solo se encuentran en los homocigotos para el alelo wild-type (genotipo CC), aunque la HbF entre estos individuos tiene una distribución muy heterogénea. Cuando el alelo mutado T está presente, tanto en heterocigosis como en homocigosis, los niveles de HbF se concentran por encima del 10%.
Para BCL11A (rs4671393), en los homocigotos para el alelo wild-type G la media ± DE de HbF fue de [13,5 ± 9,76%]; en los heterocigotos GA [14,24 ± 8,77%] y en los homocigotos para el alelo mutado A [20,09 ± 8,36%]. Los valores medios de HbF entre los genotipos de este SNP tampoco mostraron asociación estadísticamente significativa (p=0,38).
Los valores más altos de HbF entre los genotipos del SNP BCL11A (rs4671393) se reportan cuando el alelo mutado A está presente, tanto en heterocigosis GA, como en homocigosis AA, siendo bastante superiores en este último caso.
En HBS1L-MYB (rs9376092) los homocigotos para el alelo wild-type C la media ± DE de HbF fue [12,8 ± 8,06%] y en los heterocigotos CA es [14,95 ± 9,13%]. La relación de HbF entre los genotipos encontrados del SNP HBS1L-MYB (rs9376092) muestran asociación estadísticamente significativa en la población de estudio (p<0,05). No se ha encontrado ningún homocigoto para el alelo mutado.
La distribución de HbF entre los homocigotos para el alelo wild-type (genotipo CC) y los heterocigotos (genotipo CA), es diferente, siendo más elevados en esta última situación.
El número y frecuencia de SNPs de cada haplotipo se encuentra recogida en la tabla 4.
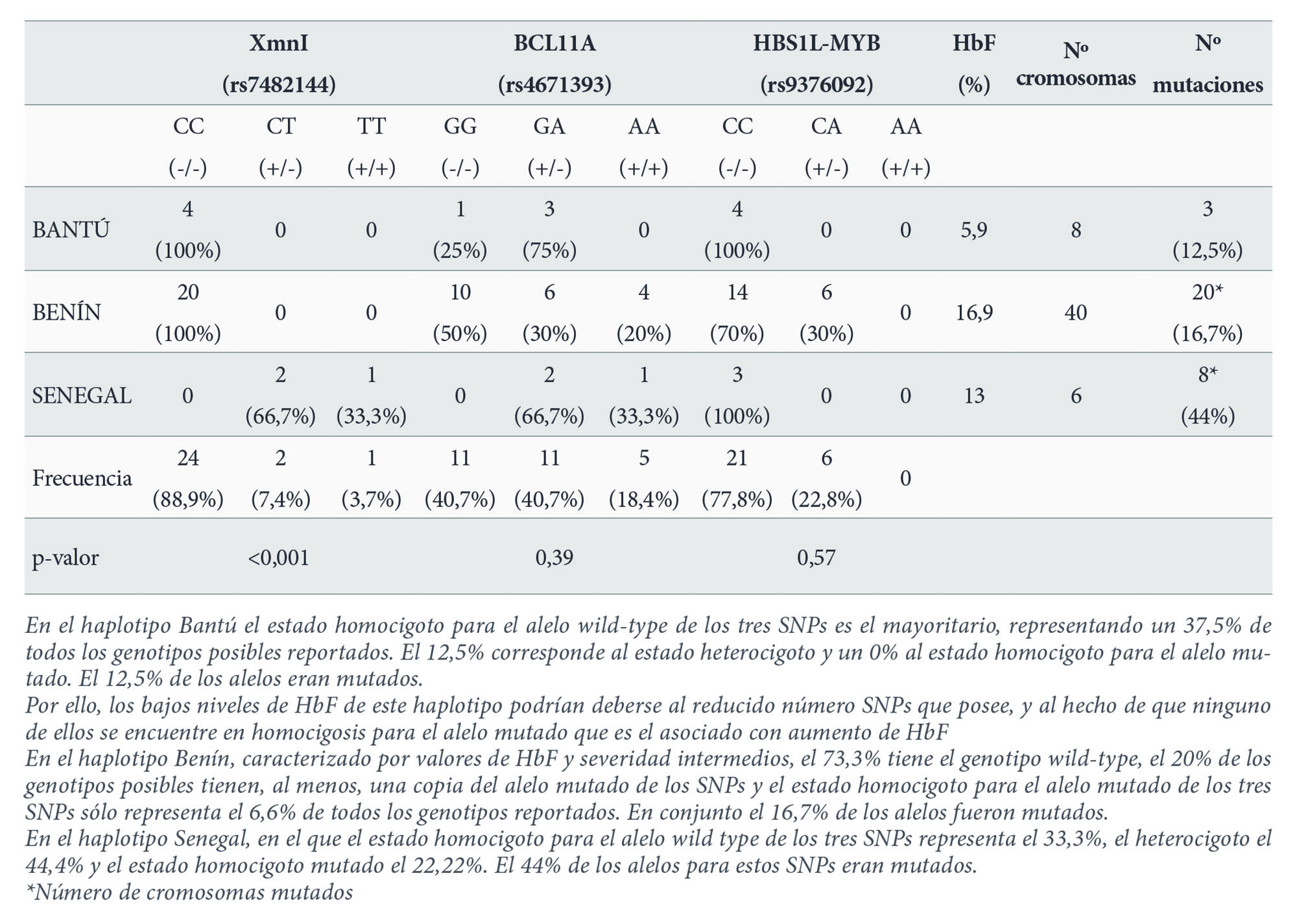
La distribución de los SNPs en BCL11A y HBS1L-MYB entre los distintos haplotipos no obtuvo resultados estadísticamente significativos; sin embargo, sí se alcanzó la asociación estadísticamente significativa con el SNP XmnI (rs7482144) (p<0,05).
La distribución de la HbF entre los SNPS en los haplotipos se ilustra en la Figura 2.
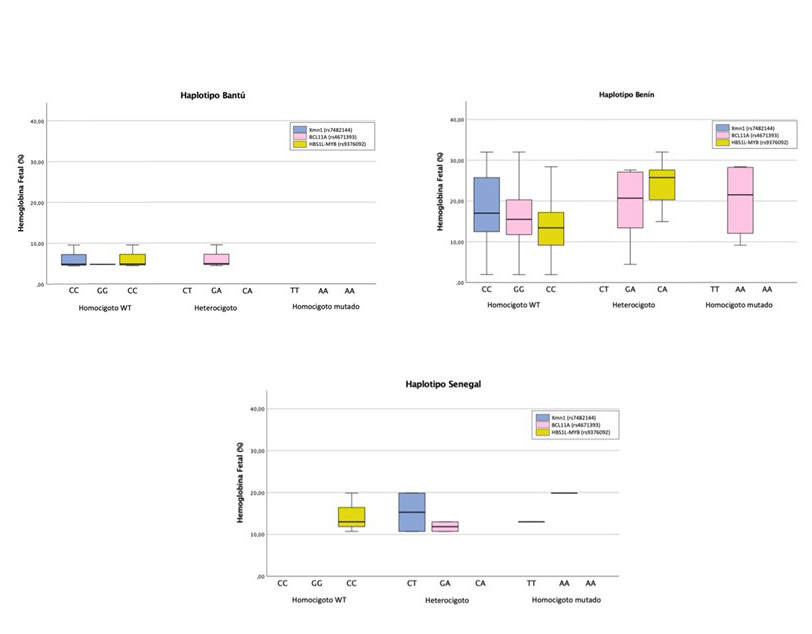
En el haplotipo Benín, el estado homocigoto para el alelo wild-type es el mayoritario en los tres SNPs, representando el 100%, el 47,4% y el 68,4% de XmnI (rs7482144), BCL11A (rs4671393) y HBS1L-MYB (rs9376092), respectivamente. Sólo se reporta el estado homocigoto para el alelo mutado del SNP BCL11A (rs4671393). Los niveles de HbF son altamente heterogéneos.
El haplotipo Senegal sólo presenta el estado homocigoto para el alelo wild-type del SNP HBS1L-MYB (rs9376092). El estado heterocigoto se encuentra en el 66,7% de los casos tanto del SNP XmnI (rs7482144), como del BCL11A (rs4671393). El genotipo homocigoto para el alelo mutado de ambos SNPs representa el 33,3%. Los niveles de HbF se concentran en la franja del 10-20%.
DISCUSIÓN
La ECF ha sido desde hace muchos años objeto de estudio por la sorprendente e inesperada heterogeneidad fenotípica de una enfermedad causada por una única mutación.
Varios factores se han relacionado con una mejoría de las complicaciones y gravedad de la enfermedad, siendo el principal y el más estudiado los niveles de HbF, los cuales también están sujetos a alta heterogeneidad, aunque existen otros como la herencia simultánea de α-talasemia o factores ambientales.
Durante la mayor parte del tiempo, la ECF ha estado restringida principalmente al África subsahariana, donde se producen cerca del 80% de los nacimientos cada año (5); sin embargo, actualmente se encuentra en la mayoría de los países debido, entre otras causas, a adopciones, migraciones por motivos económicos, políticos o conflictos bélicos, convirtiéndose en un problema de salud pública mundial (3,13).
La mayoría de los tratamientos se basan en intentar aumentar los niveles de HbF con el objetivo de conseguir un fenotipo más favorable, siendo la HU el principal fármaco utilizado. Aunque estos tratamientos son sólo paliativos y el único tratamiento curativo es el trasplante de precursores hematopoyéticos, desafortunadamente, la gran mayoría de los enfermos no pueden acceder a él debido a la escasez de donantes compatibles, bajas rentas y una sanidad inadecuada en los países con mayor incidencia, que suelen coincidir con países en vías de desarrollo (1).
Por ello existe un creciente interés por vislumbrar los factores genéticos responsables del aumento de los niveles de HbF y su variabilidad entre los pacientes e intentar manipularlos genética o farmacológicamente para conseguir un fenotipo menos grave de la enfermedad.
El objetivo de este estudio ha sido determinar si los haplotipos vinculados a mayores niveles de HbF y, por lo tanto a una enfermedad con menos complicaciones, presentan mayor número de SNPs descritos previamente como moduladores positivos de la síntesis de HbF. La presencia de estos SNPs podría explicar parcialmente la heterogeneidad de los niveles de HbF entre los haplotipos.
La HbF es un parámetro altamente variable entre individuos con ECF. En la cohorte de estudio los valores oscilan entre 1,9% y 32%. La mayor parte de esta variación (89%) está controlada por factores genéticos que se han ido identificando a lo largo de los últimos años, entre los que destacan los haplotipos del clúster β y SNPs en los tres QTLs estudiados (14).
En este estudio, el análisis de los haplotipos del clúster β ha mostrado 4 patrones diferentes, identificándose sólo haplotipos africanos, siendo el mayoritario el Benín, que el más frecuente en países próximos como Argelia o Túnez (15). De modo que los pacientes estudiados procedían o tenían ascendencia de países del África subsahariana. No se ha identificado ningún caso con el haplotipo Árabe-Indio, el cual se encuentra más restringido a la población saudí.
En la población de estudio, los individuos con el haplotipo Bantú presentaron los niveles de HbF más bajos, aproximadamente del 5%, mientras que los individuos con los niveles más altos fueron Benín y Senegal, alcanzando aproximadamente el 15%.
Los valores de HbF en los haplotipos Bantú y Senegal concuerdan con los niveles descritos en otras poblaciones, mientras que los valores de HbF en Benín varían ampliamente de unos individuos a otros (16, 17,18). No se pudo calcular la media de HbF del haplotipo Camerún ya que sólo estaba presente en un individuo (1,85%).
En nuestra población de estudio, probablemente debido al tamaño muestral, el impacto de los haplotipos del clúster β sobre los niveles de HbF no es estadísticamente significativo.
La distribución de la HbF entre los tres genotipos del SNP XmnI (rs7482144), aunque sugiere una tendencia hacia niveles superiores cuando el alelo mutado está presente, sin embargo no mostró significación estadística, dado que el alelo mutado fue identificado únicamente en dos individuos en heterocigosis (7.4%) y en uno (3,7%) en homocigosis, la mayoría de los individuos fueron homocigotos para el alelo wild-type (88,9%).
Estudios recientes parecen indicar que existen otros SNPs dentro del clúster de β-globina que se asocian de manera más significativa con la variación de HbF que el utilizado en este estudio (19).
En cuanto a los genotipos del SNP BCL11A (rs4671393) localizado en el intrón 2 del oncogén BCL11A, se reportó mayor variabilidad que en el caso anterior. El alelo mutado estuvo presente en 11 individuos (40,7%) de manera heterocigota y en 5 (18,4%) de manera homocigota. La alta frecuencia del alelo mutado en la muestra puede ser debida a que los individuos que la componen fueran africanos o de ascendencia africana, donde la frecuencia del alelo mutado es mucho mayor que en el resto de poblaciones.
Este SNP está asociado a mayores niveles de HbF, algunos estudios lo han descrito como el más influyente, se le atribuye el 13% de la variabilidad (19-21).
En nuestro estudio, los genotipos que contienen este alelo mutado presentan niveles medios de HbF superiores respecto a los homocigotos para el alelo wild-type. Existe un claro aumento de HbF en los homocigotos para el alelo mutado, donde el nivel medio de HbF es de más del 20%; sin embargo, debido al restringido tamaño muestral, no se ha podido establecer asociación estadísticamente significativa.
Del SNP HBS1L-MYB (rs9376092) sólo se han reportado dos genotipos de los tres posibles, no encontrándose ningún homocigoto para el alelo mutado. A pesar de este obstáculo, es la única variable que presenta asociación estadísticamente significativa con los niveles de HbF.
Este SNP también se asocia con la variación de HbF en cohortes de pacientes de otras poblaciones, así como en población sana (22). Está localizado en el bloque 2 de la región intergénica HBS1L-MYB, que es donde se ha reportado la asociación más fuerte con los niveles de HbF. Diversos estudios han concluido que existen otros SNPs (rs9399137 y rs9402686) dentro de este bloque que podrían estar más vinculados con esta variabilidad de HbF (7).
También existen otros SNPs en los loci clúster β-globina, BCL11A y HBS1L-MYB relacionados con la variación en los niveles de HbF; que podrían utilizarse en futuras investigaciones y comprobar si también se encuentran con mayor frecuencia en los haplotipos con mayores niveles de HbF.
La principal hipótesis del estudio era que los haplotipos con mayores niveles de HbF deberían tener mayor número de SNPs mutados.
En el haplotipo Bantú no se encontraron individuos que tuvieran dos copias del alelo mutado para ninguno de los tres SNPs. Por ello, los bajos niveles de HbF de este haplotipo podrían deberse al reducido número SNPs que posee y al hecho de que ninguno de ellos se encuentre en homocigosis para el alelo mutado que es el asociado con aumento de HbF.
En el haplotipo Benín, caracterizado por valores de HbF y severidad intermedios, el estado homocigoto para el alelo mutado de los tres SNPs en conjunto representa el 6,7% de todos los genotipos reportados. Mientras que el haplotipo Senegal es el que presenta mayor frecuencia relativa de SNPs en estado homocigoto para el alelo mutado (22,22%) de los genotipos reportados.
En base a los resultados obtenidos y acorde a la hipótesis del estudio, se observa una relación directa entre el número de SNPs homocigotos para el alelo mutado y los haplotipos con mayores niveles de HbF. Esta distribución de SNPs podría ser responsable del aumento de los niveles de HbF.
Aunque es cierto que sólo el SNP XmnI (rs7482144) alcanzó asociación estadísticamente significativa, este resultado probablemente se deba al pequeño tamaño de la muestra, por lo que sería necesario ampliar la muestra para extraer resultados más robustos y estadísticos.
Todos estos moduladores genéticos de los niveles de HbF y por lo tanto de la clínica y severidad de la ECF, se podrían utilizar como biomarcadores para estratificar a los pacientes en base a su capacidad de producir HbF, con el objetivo de un manejo clínico y farmacológico más personalizado de acuerdo al fenotipo esperado, además podría tener implicaciones en el consejo genético y en el diagnóstico prenatal de los pacientes.
Asimismo, los SNPs genéticos descritos pueden convertirse en potenciales dianas farmacológicas para desarrollar terapias novedosas que permitan aumentar el nivel de HbF en los individuos con un fenotipo más severo e intentar mejorar su desarrollo clínico. En esta línea se están desarrollado estrategias que utilizan BCL11A como diana genética ya que es un represor de la síntesis de cadenas γ-globina. Una de ellas es el silenciamiento de este gen, se ha comprobado que, silenciándolo, se consigue aumentar la producción de HbF y se corrige el fenotipo de la enfermedad en modelos de ratones de ECF, sin afectar a la eritropoyesis o a la expresión de otros genes (23). Otras estrategias se basan en la interferencia con los enhancers de BCL11A mediante ingeniería genética, para disminuir su síntesis y potenciar así la producción de HbF (24).
Se estima que la carga global de la enfermedad aumentará en los próximos años, debido a la mejora del tratamiento y a la migración hacia países con rentas más altas que permiten una supervivencia aumentada de los pacientes (5). Estas estimaciones subrayan la importancia de encontrar y explotar dianas genéticas o farmacológicas que mejoren la calidad de vida y disminuyan la mortalidad de los pacientes con ECF.
La alta heterogeneidad de esta enfermedad, aún no totalmente explicada por los factores genéticos descritos, indican que aún habría genes adicionales involucrados en la producción de HbF por descubrir, por lo que en un futuro tendremos una mayor comprensión de los mecanismos genéticos subyacentes a esta enfermedad y nuevos genes candidatos para estudio.
De cualquier manera, los avances que se están realizando permiten mejorar la calidad de vida de los pacientes y cada día estamos más cerca de conseguir una terapia personalizada que se ajuste a las características propias de cada individuo.
CONCLUSIONES
Se confirma que aquellos pacientes que tienen mayor tasa de HbF presentan un fenotipo menos severo.
La presencia del alelo mutado de cada SNP se relaciona con una tendencia hacia el aumento de HbF.
Se ha encontrado una relación directa entre los haplotipos y el número de SNPs, reportándose que los haplotipos con niveles más elevados de HbF y con ello un fenotipo menos severo, presentaban mayor número de SNPs. De modo que los haplotipos Benín y Bantú asociados tradicionalmente con mal pronóstico son los que han mostrado un menor número de SNPs mutados.
Se requieren estudios más robustos con mayor número de pacientes para confirmar nuestros hallazgos, dado el escaso o nulo porcentaje de pacientes con los haplotipos Árabe-Indio o Camerún.
DECLARACIÓN DE TRANSPARENCIA
Los autore/as de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
BIBLIOGRAFÍA
- Rees DC, Williams TN, Gladwin MT. Sickle‐cell disease. Lancet. 2010;376(9757):2018‐2031. doi: 10.1016/S0140-6736(10)61029-X
- Epstein FH, Bunn HF. Pathogenesis and Treatment of Sickle Cell Disease. N Engl J Med. 1997;337(11):762-769. doi: 10.1056/NEJM199709113371107
- Weatherall D, Akinyanju O, Fucharoen S, Olivieri N, Musgrove P, Jamison DT et al. Inherited Disorders of Hemoglobin. En: Disease Control Priorities in Developing Countries. 2º ed (2006) pp 663-680. Oxford University Press
- Rodríguez Romero WE, Sáenz Renauld GF, Chaves Villalobos MA. Hemoglobin S haplotypes: their epidemiologic, anthropologic and clinical importance]. Rev Panam Salud Publica. 1998;3(1):1-8. doi: 10.1590/s1020-49891998000100001. PMID: 9503956.
- Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010-2050: Modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013;10:e1001484
- Shenoy S. Hematopoietic stem cell transplantation for sickle cell disease: current practice and emerging trends. Hematology Am Soc Hematol Educ Program. 2011;2011:273-279. doi: 10.1182/asheducation-2011.1.273. PMID: 22160045
- Driss A, Asare KO, Hibbert JM, Gee BE, Adamkiewicz TV, Stiles JK. Sickle Cell Disease in the Post Genomic Era: A Monogenic Disease with a Polygenic Phenotype. Genomics Insights. 2009; 2009(2):23-48.
- Timofeev N, Sebastiani P, Hartley SH, Baldwin CT, Steinberg MH. Fetal Hemoglobin in Sickle Cell Anemia: A Genome-Wide Association Study of the Response to Hydroxyurea. Blood 2008; 112 (11): 2471. doi: https://doi.org/10.1182/blood.V112.11.2471.2471
- McDade J, Flanagan JM, Mortier N, et al. Genetic Predictors of Hydroxyurea Response in Children with Sickle Cell Disease. Blood 2009; 114 (22):820. doi:10.1182/blood.V114.22.820.820
- Thein SL, Menzel S, Lathrop M, Garner C. Control of fetal hemoglobin: new insights emerging from genomics and clinical implications. Hum Mol Genet. 2009;18(R2):R216-23. doi: 10.1093/hmg/ddp401.
- Torrejón MJ, Ortíz-Cabrera NV, M Nieto J, et al. Hb Moncloa: A new variant of haemoglobin that interferes in the quantification of Hb A1c. Clin Biochem. 2017;50(9):521-524. doi: 10.1016/j
- Rahimi Z, Karimi M, Haghshenass M, Merat A. Beta-globin gene cluster haplotypes in sickle cell patients from southwest Iran. Am J Hematol. 2003; 74(3):156-160. doi: 10.1002/ajh.10422. PMID: 14587041
- Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem Bulletin of the World Health Organization, 2001, 79 (8):704-712.
- Menzel S, Garner C, Gut I, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet. 2007, 39(10):1197-1199. doi: 10.1038/ng2108. 67159.
- Al-Saqladi AW, Brabin BJ, Bin-Gadeem HA, Kanhai WA, Phylipsen M, Harteveld CL. Beta-globin gene cluster haplotypes in Yemeni children with sickle cell disease. Acta Haematol. 2010;123(3):182-185. doi: 10.1159/000294965.
- Fong C, Barreto G. Presence of Non-African Haplotypes Increase Genetic Diversity in Sickle Cell Anemia Patients in Colombia. Acta Biol. Colomb, 2018; 23(3): 253-262. oi.org/10.15446/abc.v23n3.67245
- Labie D, Pagnier J, Lapoumeroulie C, Rouabhi F, et al. Common haplotype dependency of high G gamma-globin gene expression and high Hb F levels in beta-thalassemia and sickle cell anemia patients. Proc Natl Acad Sci U S A. 1985; 82(7): 2111-2114. doi: 10.1073/pnas.82.7.2111.
- Akinsheye I, Alsultan A, Solovieff N, et al. Fetal hemoglobin in sickle cell anemia. Blood. 2011;118(1): 19-27. doi: 10.1182/blood-2011-03-325258.
- Galarneau G, Palmer CD, Sankaran VG, Orkin SH, Hirschhorn JN, Lettre G. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet. 2010;42(12): 1049-1051. doi: 10.1038/ng.707.
- Uda M, Galanello R, Sanna S, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci U S A. 2008;105(5): 1620-1625. doi: 10.1073/pnas.0711566105.
- Lettre G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci U S A. 2008;105(33): 11869-11874. doi: 10.1073/pnas.0804799105. 5
- Creary LE, Ulug P, Menzel S, et al. Genetic variation on chromosome 6 influences F cell levels in healthy individuals of African descent and HbF levels in sickle cell patients. PLoS One. 2009;4(1):e4218. doi: 10.1371/journal.pone.0004218
- Xu J, Peng C, Sankaran VG, et al. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science. 2011;334(6058): 993-996. doi: 10.1126/science.1211053
- Bauer DE, Kamran SC, Lessard S, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342(6155): 253-257. doi: 10.1126/science.1242088
Paloma Ropero
Servicio de Hematología. Hospital Clínico San Carlos
C/Profesor Martín Lagos s/n · 28040 Madrid (Spain)
Tlf.: +34 913 303 321 | E-Mail: paloma.ropero@salud.madrid.org
An RANM. 2022;139(03): 285-293
Enviado: 12.11.22
Revisado: 15.11.22
Aceptado: 24.11.22