

Resumen
La Enfermedad de Células Falciformes (ECF) es una de las afecciones autosómicas recesivas más prevalentes, afectando a más de 600.000 recién nacidos al año a nivel global. En España, en el Registro Español de Hemoglobinopatías (REHem) hay inscritos 1142 casos. A pesar de la mejora de los tratamientos, la profilaxis y las vacunas, sigue siendo una enfermedad crónica y requiere cuidados de por vida. El trasplante alogénico es la única cura, siempre y cuando exista un donante HLA compatible.
La ECF es considerada, en algunas poblaciones una enfermedad rara, originada por la mutación de un solo nucleótido en el codón 6 GAG>GTG (HBB: c.20A>T) del gen β globina, genera la hemoglobina S (HbS), alterando la forma de los glóbulos rojos y obstruyendo la circulación, originando complicaciones graves en múltiples órganos. Las personas heterocigotas (βS/βA) tienen el “rasgo falciforme”, generalmente asintomático. Los homocigotos (βS/βS) presentan una enfermedad sintomática, con infartos crónicos y agudos en órganos y tejidos, episodios de dolor intenso, infartos cerebrales, esplenomegalia, episodios hemolíticos masivos y síndrome torácico agudo, con riesgo de muerte prematura.
El diagnóstico prenatal ofrece a parejas embarazadas información sobre la salud genética del feto, permitiéndoles tomar decisiones informadas. Aunque las técnicas convencionales son invasivas y conllevan riesgos, se ha desarrollado el Diagnóstico Prenatal No Invasivo (DPNI) usando ADN libre fetal circulante (cffDNA) en la sangre materna para determinar el genotipo del feto. Sin embargo, la presencia conjunta de ADN materno y ADN fetal, y la baja concentración de este último, ha supuesto un desafío en los estudios no invasivos.
Presentamos los resultados de un estudio piloto donde se ha establecido el uso de la PCR digital (dPCR) para el DPNI de la ECF, una técnica altamente sensible que permite detectar mutaciones en muestras de baja frecuencia, presentándose como una alternativa segura para el diagnóstico de la ECF.Abstract
Sickle Cell Disease (SCD) is one of the most prevalent autosomal recessive disease, affecting over 600,000 newborns globally each year. In Spain, the Spanish Registry of Hemoglobinopathies (REHem) has registered 1142 cases. Despite improvements in treatments, prophylaxis, and vaccines, it remains a chronic condition requiring lifelong care. Allogeneic transplantation is the only cure, contingent upon the availability of an HLA-compatible donor.
SCD is considered a rare disease in some population, resulting from a mutation of a single nucleotide in codon 6 GAG>GTG (HBB: c.20A>T) of the β-globin gene, producing hemoglobin S (HbS), altering the shape of red blood cells and obstructing circulation, leading to severe complications in multiple organs. Heterozygous individuals (βS/βA) have the ‘sickle cell trait,’ typically asymptomatic. Homozygotes experience symptomatic disease, with chronic and acute infarctions in organs and tissues, episodes of intense pain, cerebral infarctions, splenomegaly, massive hemolytic episodes, and acute chest syndrome, causing a risk of premature death increasing the threat of premature mortality.
Prenatal diagnosis provides expecting couples with information about the fetus’s genetic health, enabling informed decision-making. Although conventional techniques are invasive and carry risks, Non-Invasive Prenatal Diagnosis (NIPD) has been developed using cell free fetal DNA (cffDNA) in maternal blood to determine the fetus’s genotype. However, the co-presence of maternal DNA and cffDNA, along with the low concentration of the latter, has been a challenge in non-invasive studies.
We present the results of a pilot study where the use of digital PCR (dPCR) has been established for NIPD of SCD, a highly sensitive technique allowing the detection of mutations in low-frequency samples, presenting itself as a safe alternative for SCD diagnosis.Palabras clave: Enfermedad células falciforme (ECF); Hemoglobina S (HbS); Diagnóstico prenatal no invasivo (DPNI); PCR digital (dPCR).
Keywords: Sickle cell disease (SCD); Hemoglobin S (HbS); Non-invasive Prenatal diagnosis (NIPD); Digital PCR (dPCR).
INTRODUCCIÓN
La Enfermedad de Células Falciformes (ECF) es una afección crónica que se caracteriza por la presencia de anemia hemolítica de intensidad moderada a grave, que suele manifestarse a partir del tercer o cuarto mes de vida. Además, conlleva una vasculopatía que da lugar a fenómenos de vasooclusión, provocando isquemia y daño crónico en varios órganos. Esta patología se ve interrumpida por episodios agudos, y los pacientes presentan una notable susceptibilidad a infecciones debido a la falta de funcionamiento del bazo, así como episodios de hemólisis masiva y el riesgo de síndrome torácico agudo, que puede llevar a la muerte prematura (1).
La ECF es ocasionada por una mutación en un solo nucleótido en el codón 6 (GAG>GTG) del gen β-globina (HBB: c.20A>T) (NM_000518.4). Esta mutación resulta en la producción de la hemoglobina S (HbS), caracterizada por la sustitución de un único aminoácido, donde Glu se convierte en Val en la posición β6(A3) (2). Este cambio permite que la HbS forme polímeros cuando está desoxigenada, lo que distorsiona la morfología de los glóbulos rojos, adoptando una característica forma de hoz que obstruye la circulación microvascular, desencadenando los síntomas clínicos mencionados previamente.
Según los datos epidemiológicos actuales, aproximadamente el 7% de la población mundial porta un gen de hemoglobina anormal, y cada año nacen más de 600,000 niños afectados por esta enfermedad. La gran mayoría de los casos de ECF (más del 83%) se deben a la alta prevalencia del gen de la drepanocitosis en regiones específicas, como Oriente Medio, Asia Meridional, el Mediterráneo y el África subsahariana. Las migraciones de población y los matrimonios mixtos entre diferentes grupos étnicos han introducido la ECF en prácticamente todos los países del mundo, independientemente de si son endémicos de la malaria o no, incluyendo el norte de Europa y otros lugares donde estas condiciones no existían previamente (3).
En España, de acuerdo con los datos del Registro Español de Hemoglobinopatías (REHem), hay inscritos 1.142 casos de ECF, y se ha observado un aumento progresivo de casos de anemia falciforme debido a los flujos migratorios procedentes del Caribe, Sudamérica y África. La detección temprana, el tratamiento profiláctico con penicilina y las vacunas han contribuido a mejorar la calidad de vida y aumentar la esperanza de vida de los pacientes con ECF. Sin embargo, esta sigue siendo una afección debilitante que requiere atención a lo largo de toda la vida, y el único tratamiento curativo es el trasplante alogénico, siempre que exista un donante compatible HLA (4).
El diagnóstico prenatal de enfermedades brinda a las parejas embarazadas la oportunidad de obtener información precisa y rápida sobre la salud genética de su feto. Esto les permite tomar decisiones fundamentadas, ya sea considerar la interrupción del embarazo en caso de obtener resultados positivos o prepararse tanto emocional como médicamente para la llegada de su hijo cuando el aborto no es una opción. Aunque las técnicas convencionales, como la biopsia corial (CVS) o la amniocentesis, son efectivas, son invasivas y conllevan riesgos para el feto, aunque en menor medida para la madre (5,6,7).
Para abordar estos desafíos, se ha desarrollado el Diagnóstico Prenatal No Invasivo (DPNI), basado en el uso de ADN libre fetal (cell free fetal DNA – cffDNA) presente en la sangre materna procedente de las células trofoblásticas de la placenta (8,9,10). El ADN fetal varía en concentración a medida que avanza el embarazo y en respuesta a factores maternos o fetales. En las últimas dos décadas, se ha avanzado significativamente en la comprensión del ADN libre fetal, ya que su presencia nos permite obtener información genética del feto a partir de sangre materna (11, 12).
A pesar de estos avances, el DPNI se enfrenta a desafíos debido a la coexistencia del ADN materno y del fetal, así como a la baja concentración de ADN libre fetal en la sangre materna, lo que ha dado lugar a falsos negativos y diagnósticos incorrectos en algunos casos. Sin embargo, el DPNI se ha convertido en una parte integral de la práctica clínica y se utiliza para determinar el sexo fetal (13), evaluar el factor RhD (14), realizar el cribado de aneuploidías comunes y diagnosticar mutaciones paternas o de novo (15).
Para enfermedades monogénicas, la aplicación del DPNI ha estado limitada a aquellas con herencia paterna y de novo debido a la mayor complejidad de distinguir los alelos maternos de los fetales. En estos casos, las técnicas de análisis varían según la base genética subyacente, incluyendo técnicas como la PCR Cuantitativa Fluorescente (QF-PCR), PCR en tiempo real (RT-PCR) y Minisecuenciación o SNaPshot (16).
No obstante, para enfermedades en las cuales ambos progenitores poseen la misma mutación, el DPNI no se basa en el principio de presencia/ausencia de la mutación, inútil en este contexto ya que la madre presenta el alelo wildtype y mutado y no se puede determinar si la mutación observada es materna o fetal, sino en el estudio de la dosis relativa mutacional (RMD) (12, 16), llevado a cabo mediante PCR digital (dPCR). La dPCR permite una cuantificación absoluta de los alelos wild type y mutado de forma altamente sensible y específica (17). Esta cuantificación permite conocer si existe equilibrio o desequilibrio alélico entre los alelos normal y mutado en la muestra observada, esperando que una mujer sin transfusiones ni embarazada se encuentre en equilibrio alélico, 50:50 entre los alelos wildtype y mutado. Esto unido a la presencia de ADN fetal en plasma materno, resulta en que pueda darse un desequilibrio dado por la contribución fetal al ADN total, de forma que si la madre y el feto comparten el mismo genotipo se mantiene el equilibrio alélico; mientras que si el feto es homocigoto habrá un aumento del alelo del que sea homocigoto el feto (18).
Presentamos los resultados preliminares de un estudio piloto en proceso de validación de un método de DPNI para la detección de la ECF mediante dPCR.
MATERIAL Y MÉTODOS
Se llevó a cabo un estudio observacional con intervención diagnóstica que incluyó a 9 mujeres embarazadas con rasgo falciforme (βA/βS) y mujeres no embarazadas con los tres posibles genotipos: 6 homocigotas wildtype (βA/βA), 8 heterocigotas (βA/βS), y 6 homocigotas para la mutación (βS/βS).
Se excluyeron aquellas mujeres que recientemente hubieran recibido transfusiones sanguíneas, trasplantes alogénicos, terapias inmunológicas o de células madre debido a posibles interferencias por ADN exógeno y gestaciones múltiples.
Las muestras fueron reclutadas en varios hospitales de Madrid, Clínico San Carlos, Universitario de Getafe, Universitario Príncipe de Asturias y en los hospitales Universitario de Cruces (Bilbao), Universitario de Guadalajara, Universitario Marqués de Valdecilla (Santander), Clínico Universitario Virgen de la Arrixaca (Murcia), General de Segovia y Universitario Miguel Servet (Zaragoza).
Las muestras fueron procesadas y caracterizadas molecularmente en el laboratorio de Eritropatología del servicio de Hematología del Hospital Clínico San Carlos. Se realizó un estudio hematimétrico convencional y un recuento de reticulocitos utilizando un contador automático de células (UniCel DxH 800, Coulter Cellular Analysis System, Beckman Coulter). La identificación y cuantificación de la HbS y las distintas fracciones de hemoglobina se llevaron a cabo mediante HPLC de intercambio iónico (Variant II, BioRad) y electroforesis capilar (Minicap Flex Piercing, Sebia).
Para el análisis molecular, se extrajo el ADN de sangre periférica mediante extracción automática de los leucocitos (EZ1, Qiagen) y se caracterizó empleando el kit comercial β-Globin StripAssay® MED (ViennLab). La obtención del ADN libre circulante se realizó recolectando sangre periférica entre la semana 10 y 20 de gestación en tubos K3EDTA Cell-free DNA BCT (Streck). Posteriormente, se extrajo el ADN libre circulante utilizando el kit comercial QIAamp Circulating Nucleic Acid kit (Qiagen).
La caracterización molecular del ADN libre circulante se llevó a cabo mediante dPCR [QuantStudio 3D Digital PCR System (Thermo Fisher)] con sondas TaqMan específicas para el alelo mutado (FAM) y el alelo wildtype (VIC) (Tabla 1). Esto permitió una cuantificación absoluta de ambos alelos mediante el software QuantStudio 3D AnalysisSuite Cloud Software (ThermoFisher) y determinar el RMD de la mutación (HBB: c.20A>T) mediante la siguiente fórmula:

RMD = [Nº eventos positivos marcados con FAM (mutado)]/(Nº de eventos positivos totales VIC +FAM).
Los valores de RMD obtenidos se tradujeron en valores de z-score utilizando una fórmula (19) basada en una población de referencia de mujeres heterocigotas no embarazadas, estableciendo rangos de equilibrio/desequilibrio para asociar al feto un genotipo determinado.
z-score = (x – μ) / σ
donde:
χ = RMD obtenido
μ = media de la población de referencia
σ = desviación estándar de la población de referencia
Se determinaron puntos de corte para clasificar con un 99% de confianza los genotipos fetales. Los valores de z-score inferiores a -2,6 se asociaron con mujeres embarazadas con feto homocigoto wildtype, mientras que aquellos superiores a +2,6 indicaron mujeres embarazadas con feto homocigoto mutado. Los valores entre -1,5 y +1,5 se asignaron a mujeres embarazadas con feto heterocigoto. Además, se mantuvo una zona intermedia donde el resultado se consideró no concluyente.
Una vez obtenido el diagnóstico en las gestantes, se verificó el resultado. Para ello se realizó el diagnóstico de los recién nacidos, mediante el cribado neonatal de las hemoglobinopatías por la prueba del talón, que se lleva a cabo de forma universal en todo el territorio nacional.
En el análisis estadístico descriptivo de los datos, las variables cualitativas se presentan con su distribución de frecuencia. Las variables cuantitativas se resumen con su media y desviación estándar (DE). Las variables cuantitativas que muestran una distribución asimétrica se resumen con la mediana y el rango intercuartílico (RIC). Se utilizó el z-score para clasificar los pacientes en el grupo de embarazadas portadoras de HbS.
El procesamiento de los datos se llevó a cabo mediante el software estadístico IBM SPSS Statistics v.26 y Microsoft Excel.
El estudio se realizó con el consentimiento informado de todas las participantes y cuenta con la aprobación del CEIm Hospital Clínico San Carlos de Madrid.
RESULTADOS
Análisis de RMD utilizando PCR digital
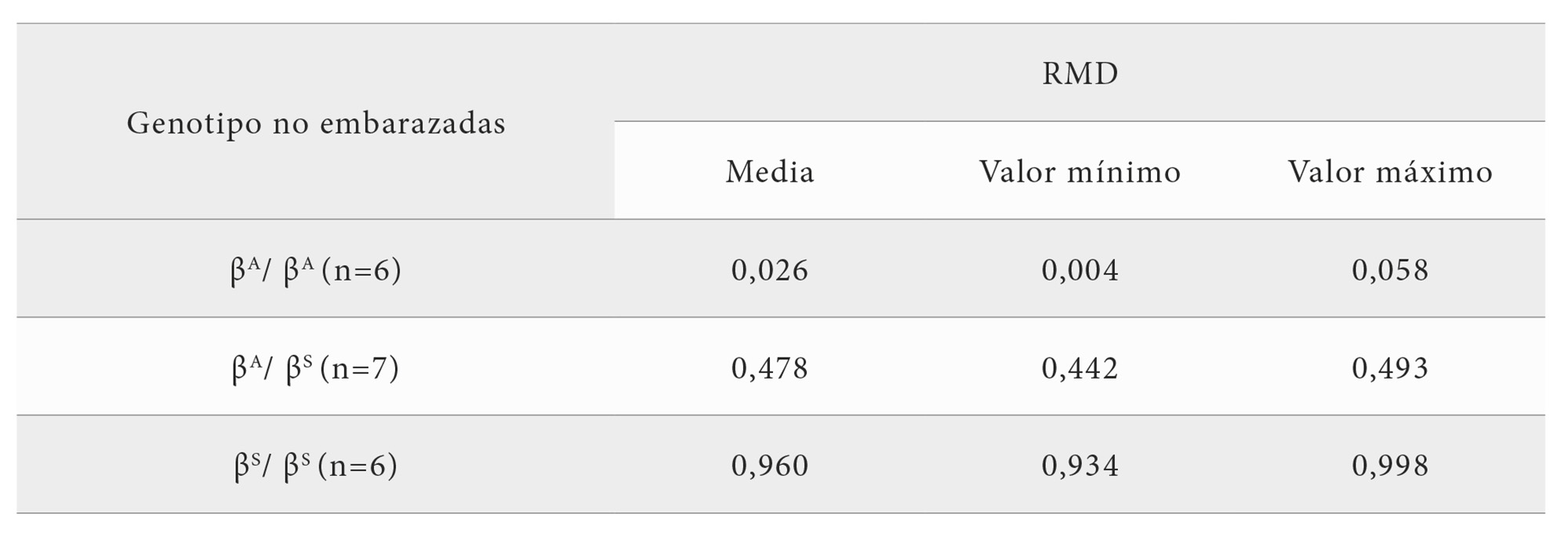
Se implementó la técnica de dPCR para determinar la RMD de alelos mutados en diferentes grupos de estudio. Los valores de RMD obtenidos demostraron una clara diferenciación entre las mujeres no embarazadas con distintos genotipos para la mutación HBB: c.20A>T. Aquellas que presentaban un genotipo homocigoto para la mutación, los valores de RMD se aproximaron a 1, con una media de 0.96, mientras que en las que el genotipo era wildtype, los valores de RMD estuvieron cercanos a 0, con una media de 0,02. Por último, en las heterocigotas se mantuvo el equilibrio alélico, y se observaron valores de RMD que oscilaron entre 0,442 y 0,493, con una media de 0,478 (Figura 1) (Tabla2).
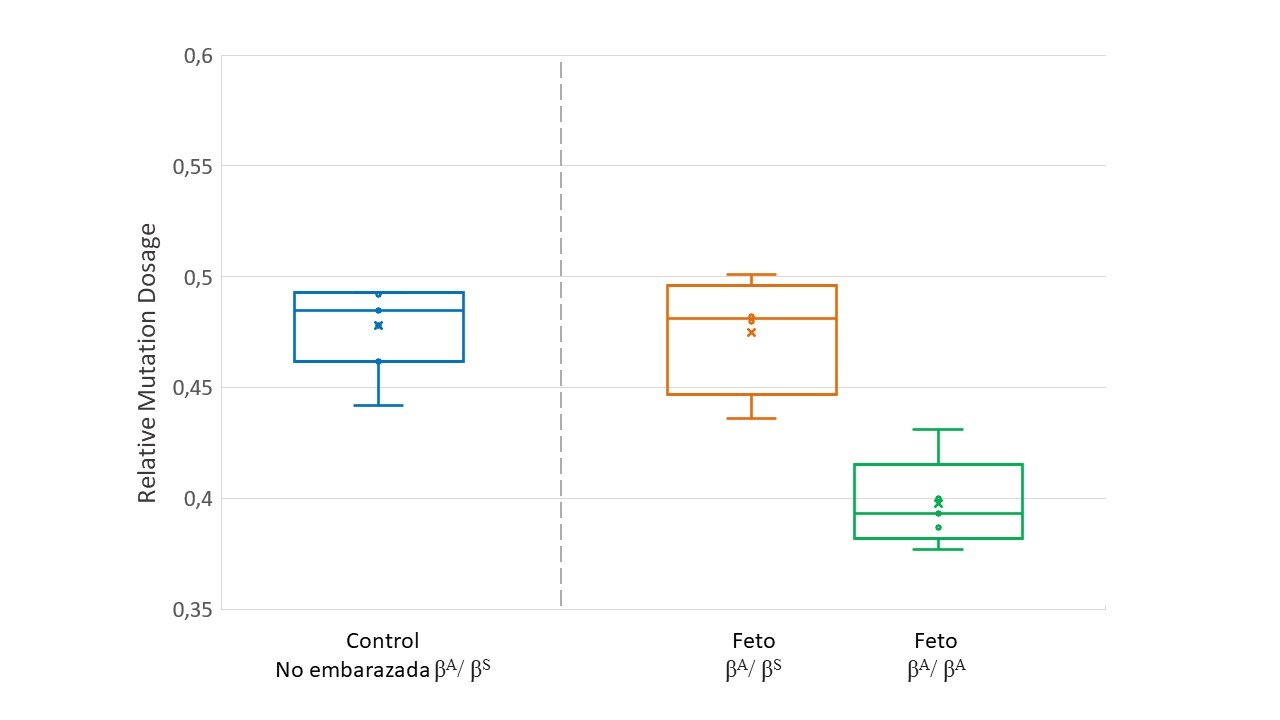
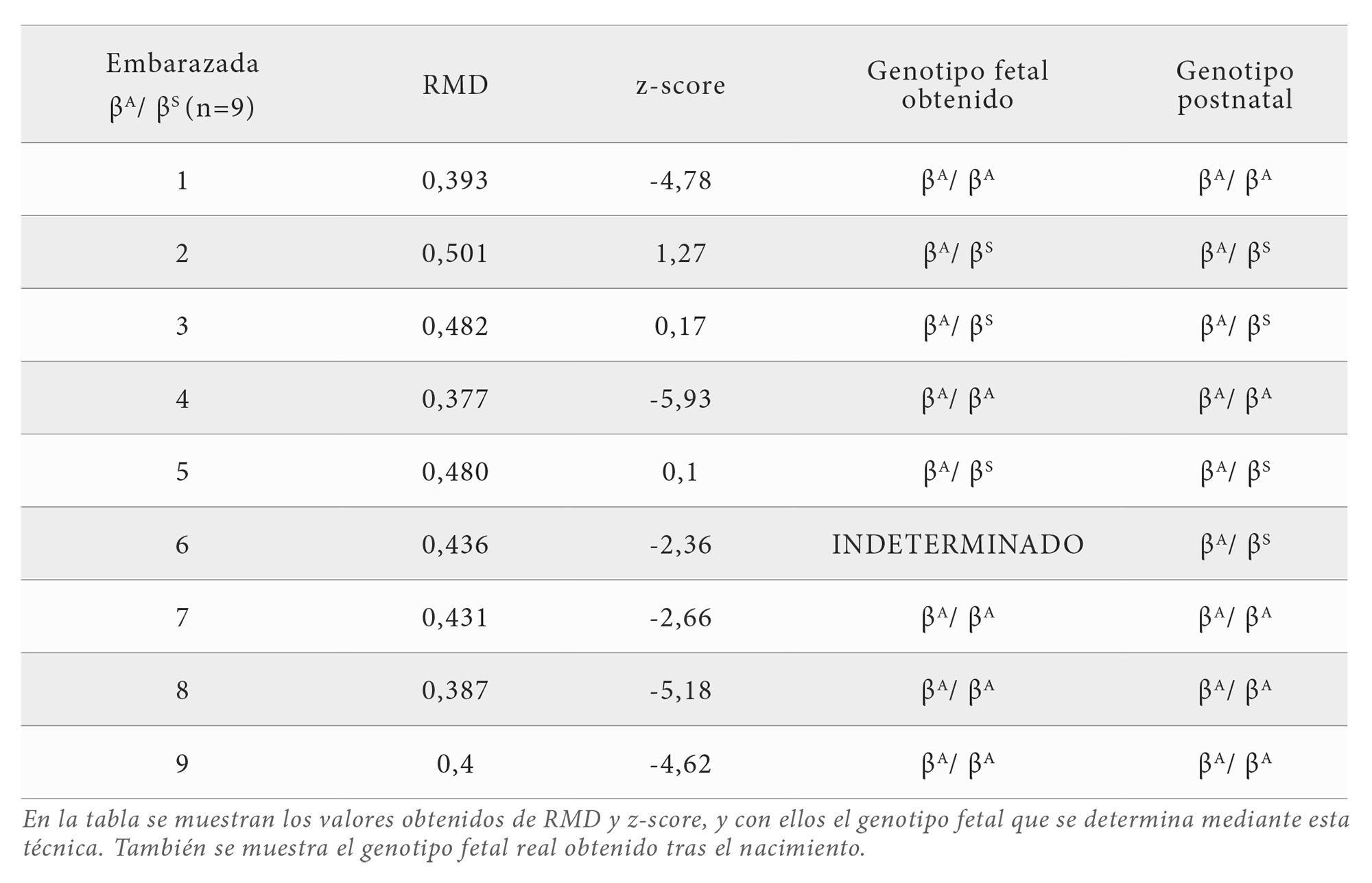
En el grupo de mujeres embarazadas, todas ellas portadoras de ECF, se detectaron resultados distintivos en función del genotipo fetal. Los resultados obtenidos mostraron variaciones en los valores de RMD que permitieron diferenciar eficazmente entre feto heterocigoto y feto wildtype. Específicamente, en mujeres con feto heterocigoto, los valores de RMD se mantuvieron entre 0,436 y 0,501, con una media de 0,475. Por otro lado, en mujeres con feto homocigoto wildtype, los valores de RMD fluctuaron entre 0,377 y 0,431, con una media de 0,397 (Figura 1) (Tabla 3).
Determinación del genotipo fetal mediante z-score
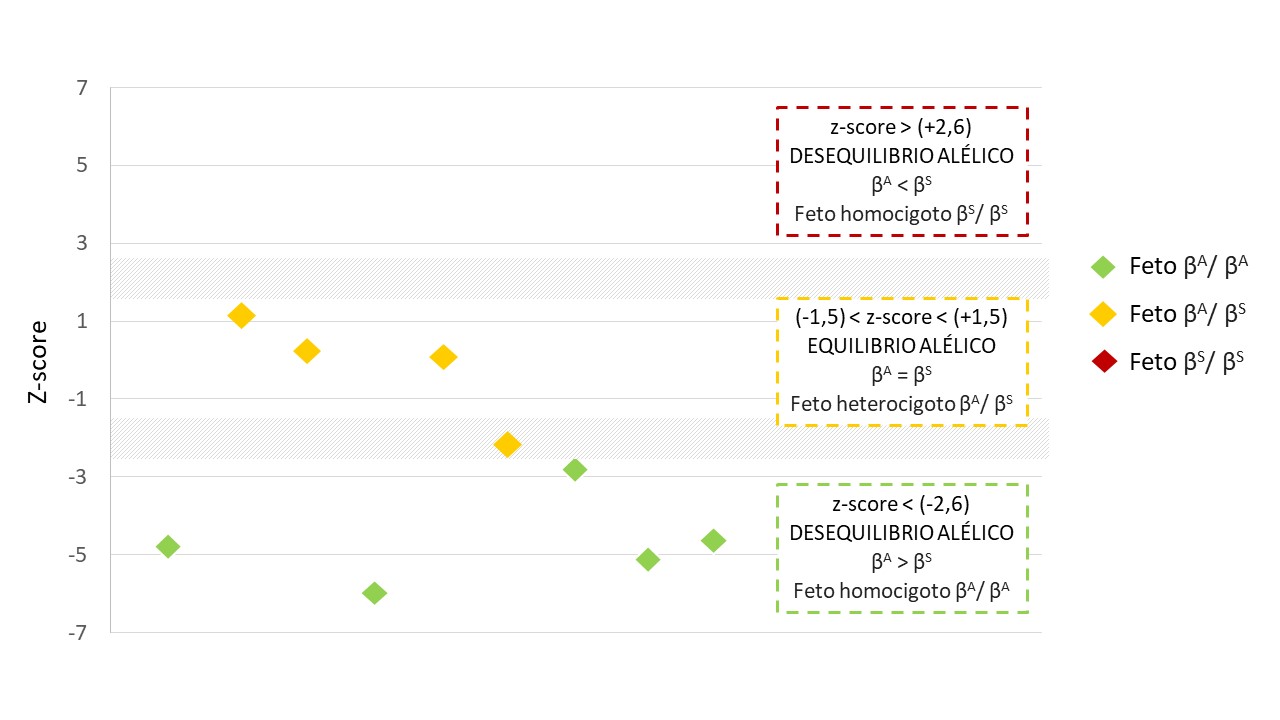
Los resultados obtenidos de la caracterización genética revelaron una distribución interesante en relación con los valores obtenidos. Se identificaron 5 resultados que se ubicaron en una zona verde, con valores comprendidos entre -5,93 y -2,66 en la escala de referencia utilizada. Estos resultados indican un rango específico de valores z-score, que podría estar asociado con ciertos parámetros de normalidad o equilibrio en el contexto genético analizado. Además, se detectaron 3 resultados que se posicionaron dentro de una zona amarilla, presentando valores z-score que oscilaron entre 1,27 y 0,1. Esta zona amarilla representa otro espectro de valores que podrían sugerir un cierto grado de desequilibrio o variación en comparación con el estándar de referencia establecido. Un hallazgo particular fue la identificación de un único resultado en una zona gris, el cual mostró un valor de -2,36 en la escala de z-score. Esta zona gris podría indicar un punto intermedio o una situación particular que requiere un análisis más detallado, dado su alejamiento de los rangos convencionales establecidos en la referencia.
Estos hallazgos se visualizan de manera clara y precisa en la Figura 2, donde se sitúan los valores z-score de los resultados obtenidos. Esta representación gráfica facilita la comprensión y la visualización de la distribución de los valores obtenidos en cada zona específica, permitiendo una mejor interpretación de los datos.
Verificación y confirmación del genotipo fetal
Tras la aplicación de los puntos de corte, se verificó el genotipo fetal mediante otros métodos de diagnóstico incluyendo el cribado neonatal de las hemoglobinopatías. Esto permitió confirmar que los resultados obtenidos coincidieron con precisión en un 100% de los casos en los que se identificó el feto homocigoto wildtype y en un 100% de los casos en los que se identificó el feto heterocigoto. Sin embargo, un caso, clasificado en la zona intermedia, no pudo ser concluyente y requirió repetición del estudio y correspondió a una mujer embarazada con feto heterocigoto.
Teniendo 4 mujeres heterocigotas embarazadas con genotipo fetal heterocigoto, y habiendo clasificado con z-score el 75% en equilibrio alélico, se puede esperar que en el grupo de mujeres heterocigotos embarazadas con feto homocigoto sólo el 5% no lo clasifique como desequilibrio. Por lo que con estos datos nuestro estudio tiene una potencia del 70%.
Hasta la fecha no se han integrado casos de mujeres embarazadas con feto homocigoto mutado.
DISCUSIÓN
Hasta el momento, las enfermedades monogénicas de herencia recesiva no se han podido beneficiar del DPNI debido a la imposibilidad de separar el ADN materno y fetal, lo cual supone un desafío en la detección de variantes de herencia materna (20).
Este estudio destaca la dPCR como una técnica óptima y valiosa para la detección prenatal no invasiva (DPNI) de la anemia falciforme (ECF) en madres portadoras de la mutación (21). El análisis del RMD mediante dPCR reveló patrones distintivos entre mujeres no embarazadas heterocigotas y homocigotas para la mutación. En el grupo de mujeres embarazadas, nos basamos en el RMD y en la contribución fetal. Se espera mantener un equilibrio alélico en los casos de madre y feto heterocigotos, y observar un ligero desequilibrio en los casos en que el feto sea homocigoto (12, 18).
En los nueve casos estudiados de mujeres embarazadas portadoras de la mutación de la ECF, se observaron variaciones en los valores de RMD en función del genotipo fetal y de la cantidad de ADN fetal en plasma materno (12), lo que permitió una diferenciación efectiva entre heterocigoto y homocigoto wildtype.
Observamos valores muy similares al control en los casos de embarazadas de feto heterocigoto (media control = 0,478; media embarazada de feto heterocigoto = 0,475), indicativo de que se mantiene el equilibrio alélico. En cambio, en embarazadas con fetos homocigotos wildtype, los valores de RMD mostraron ligeras diferencias (media embarazada feto wildtype = 0,397), alejándose del equilibrio observado en el grupo control, dado por la aportación del ADN fetal, que aumenta ligeramente el número de copias del alelo wildtype y disminuyendo el RMD. Y aunque no hemos encontrado ningún caso en nuestro estudio, si la embarazada portara un feto homocigoto mutado se esperaría el caso opuesto, un pequeño desequilibrio pero que aumente el número de copias del alelo mutado aumentando el RMD, con valores cercano al 1. Por tanto, existen diferencias en la dosis relativa mutacional en función del genotipo fetal dadas por la presencia de ADN fetal en sangre materna, confirmando los resultados de Perlado y Hanson para el DPNI de enfermedades monogénicas recesivas (18, 23).
El RMD por sí sólo lo único que hace es revelar un patrón distintivo, pero que sin embargo y dado que la concentración de ADN fetal es baja, para poder diferenciar los genotipos fetales se requiere del cálculo del z-score, una medida de la diferencia entre el RMD de cada una de las embarazadas respecto al grupo control (24, 25). El valor que se obtiene de este parámetro será el que nos determine el genotipo fetal. En nuestro estudio se comprobó que todos los fetos que presentaron un z-score > -2,6 fueron correctamente clasificados como homocigotos para el alelo wildtype (5/5); también fueron correctamente clasificados aquellos que presentaron valores entre 1,3 y 0,1 como fetos heterocigotos (3/4), sólo uno no pudo ser correctamente clasificado ya que el valor que se obtuvo se situaba en la denominada zona intermedia (zona gris). Esta zona aporta seguridad en el diagnóstico evitando falsos positivos o negativos (18, 22).
Los resultados obtenidos a través de la aplicación de la técnica de dPCR para determinar el genotipo fetal en mujeres embarazadas portadoras de la mutación de la ECF ofrecen una perspectiva prometedora en el campo del DPNI. Estos resultados se alinean con investigaciones previas que han explorado métodos no invasivos para el diagnóstico de enfermedades monogénicas, enfocándose en la identificación precisa del genotipo fetal a partir de muestras de sangre materna (26).
La precisión y capacidad de diferenciación entre genotipos observada en este estudio son consistentes con las características fundamentales del análisis de la RMD mediante dPCR. Esto resalta la viabilidad de esta técnica como una herramienta valiosa para identificar de manera no invasiva el genotipo fetal en mujeres embarazadas portadoras de la mutación de la ECF. Además, estos resultados preliminares respaldan la idea de que la dPCR, al permitir la detección sensible y específica de alelos mutados, puede tener un impacto significativo en la práctica clínica al ofrecer una alternativa menos invasiva y más segura para el diagnóstico prenatal de esta enfermedad (23)
Sin embargo, es crucial considerar algunas limitaciones inherentes a este estudio piloto. La muestra utilizada fue limitada y, aunque los resultados son prometedores, se necesitan investigaciones adicionales con muestras más amplias para validar plenamente la precisión y seguridad de la técnica en la determinación del genotipo fetal en casos de ECF. Además, la falta de inclusión de casos de mujeres embarazadas con feto homocigoto mutado en este estudio limita la generalización de los resultados y destaca la necesidad de investigaciones futuras que aborden esta área específica.
A nivel clínico, los hallazgos de este estudio tienen implicaciones significativas. La capacidad de determinar el genotipo fetal de manera no invasiva puede impactar en la toma de decisiones informadas por parte de los padres, permitiendo la planificación y preparación adecuadas para el cuidado del niño afectado por la ECF cuando la interrupción del embarazo no sea una opción. Además, esta técnica podría minimizar los riesgos asociados con procedimientos invasivos de diagnóstico prenatal, como la amniocentesis o la biopsia corial, mejorando así la seguridad y bienestar tanto de la madre como del feto (5-7).
El éxito de esta investigación resalta la importancia de la continua exploración y desarrollo de herramientas de DPNI. La aplicación exitosa de la dPCR en la determinación del genotipo fetal para la ECF sugiere su potencial utilidad en otras enfermedades monogénicas hereditarias, abriendo la puerta a futuras investigaciones que exploren su aplicabilidad en una gama más amplia de condiciones genéticas.
En resumen, si bien este estudio piloto ofrece resultados alentadores en el uso de la dPCR para el DPNI de la ECF, se requiere más investigación para consolidar y validar plenamente la utilidad clínica de esta técnica en la determinación precisa del genotipo fetal en casos de esta enfermedad y su posible aplicación en otros trastornos genéticos.
AGRADECIMIENTOS
La realización de este proyecto fue posible gracias al respaldo financiero otorgado por la beca concedida por el Grupo Español de Eritropatología de la Sociedad Española de Hematología y Hemoterapia en el año 2022.
DECLARACIÓN DE TRANSPARENCIA
Los autores/as de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
BIBIOGRAFÍA
- ↑Rees DC, Williams TN, Gladwin MT. Sickle‐cell disease. Lancet. 2010; 376(9757): 2018‐2031. doi: 10.1016/S0140-6736(10)61029-X
- ↑Epstein FH, Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997; 337(11): 762-769. doi: 10.1056/NEJM199709113371107
- ↑Piel FB, Hay SI, Gupta S, Weatherall DJ, Williams TN. Global burden of sickle cell anaemia in children under five, 2010-2050: Modelling based on demographics, excess mortality, and interventions. PLoS Med. 2013; 10(7): e1001484.
- ↑Shenoy S. Hematopoietic stem cell transplantation for sickle cell disease: Current practice and 280 emerging trends. Hematology Am Soc Hematol Educ Program. 2011; 2011: 273-279. doi: 281 10.1182/asheducation-2011.1.273. PMID: 22160045
- ↑Kozlowski P, Knippel A, Stressig R. Individual risk of fetal loss following routine second trimester amniocentesis: a controlled study of 20,460 cases. Ultraschall Med. 2008; 29(2): 165-172. doi: 10.1055/s-2007-963217.
- ↑Mujezinovic F, Alfirevic Z. Procedure-related complications of amniocentesis and chorionic villous sampling: a systematic review. Obstet Gynecol. 2007; 110(3): 687-694. doi: 10.1097/01.AOG.0000278820.54029.e3. Erratum in: Obstet Gynecol. 2008;111(3):779. PMID: 17766619
- ↑Ropero P, González FA, Hernández A, Sánchez H, Cela E, Villegas A. Diagnóstico prenatal de hemoglobinopatias y talasemias. Med Clin (Barc). 2009; 132(2): 53-56.
- ↑Lo YM, Corbetta N, Chamberlain PF et al. Presence of fetal DNA in maternal plasma and serum. Lancet. 1997; 350: 485-487. doi: 10.1016/S0140-6736(97)02174-0
- ↑Alberry M, Maddocks D, Jones M et al. Free fetal DNA in maternal plasma in anembryonic pregnancies: Confirmation that the origin is the trophoblast. Prenat Diagn. 2007; 27(5): 415-418. doi: 10.1002/pd.1700
- ↑Everett TR., Chitty LS. Cell-free fetal DNA: the new tool in fetal medicine: cffDNA in fetal medicine. Ultrasound Obst Gyn. 2015; 45(5): 499-507. doi.org/10.1002/uog.14746
- ↑Lo YM, Chan KC, Sun H et al. Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med. 2010; 2(61): 61ra91. doi: 10.1126/scitranslmed.3001720. PMID: 21148127.
- ↑Lun FM, Tsui NB, Chan KC et al. Noninvasive prenatal diagnosis of monogenic diseases by digital size selection and relative mutation dosage on DNA in maternal plasma. Proc Natl Acad Sci USA. 2008; 105(50): 19920-19925. doi: 10.1073/pnas.0810373105.
- ↑Wu D, Hou Q, Li T et al. The use of cffDNA in fetal sex determination during the first trimester of pregnancy of female DMD carriers. Intractable Rare Dis Res. 2012; 1(4): 157-160. doi: 10.5582/irdr.2012.v1.4.157
- ↑Teitelbaum L, Metcalfe A, Clarke G, Parboosingh JS, Wilson RD, Johnson JM. Costs and benefits of non-invasive fetal RhD determination. Ultrasound Obst Gyn. 2015; 45(1): 84-88. doi: 10.1002/uog.14723
- ↑Gruber A, Pacault M, El Khattabi LA et al. Non-invasive prenatal diagnosis of paternally inherited disorders from maternal plasma: Detection of NF1 and CFTR mutations using droplet digital PCR. Clin Chem Lab Med. 2018; 56(5): 728-738. doi: 10.1515/cclm-2017-0689
- ↑Rodríguez de Alba M, Bustamante-Aragonés A, Perlado S et al. Noninvasive prenatal diagnosis of monogenic disorders. Expert Opin Biol Ther. 2012; 12(Suppl1): S171-S179. doi: 10.1517/14712598.2012.674509.
- ↑Mao X, Liu C, Tong H, Chen Y, Liu K. Principles of digital PCR and its applications in current obstetrical and gynecological diseases. Am J Transl Res. 2019; 11(12): 7209-7222. eCollection 2019.
- ↑Hanson B, Scotchman E, Chitty LS, Chandler NJ. Non-invasive prenatal diagnosis (NIPD): How analysis of cell-free DNA in maternal plasma has changed prenatal diagnosis for monogenic disorders. Clin Sci (Lond). 2022; 136(22): 1615-1629. doi: 10.1042/CS20210380
- ↑Andrade C. Z Scores, standard scores, and composite test scores explained. Indian J Psychol Med. 2021; 43(6): 555-557. doi: 10.1177/02537176211046525
- ↑Lench N, Barrett A, Fielding S et al. The clinical implementation of non-invasive prenatal diagnosis for single-gene disorders: Challenges and progress made. Prenat Diagn. 2013; 33(6): 555-562. doi: 10.1002/pd.4124.
- ↑Nectoux J. Current, Emerging, and future applications of digital PCR in non-invasive prenatal diagnosis. Mol Diagn Ther. 2018; 22(2): 139-148. doi: 10.1007/s40291-017-0312-x.
- ↑Barrett AN, McDonnell TC, Chan KC, Chitty LS. Digital PCR analysis of maternal plasma for noninvasive detection of sickle cell anemia. Clin Chem. 2012; 58(6): 1026-1032. doi: 10.1373/clinchem.2011.178939.
- ↑Perlado S, Bustamante-Aragonés A, Donas M, Lorda-Sánchez I, Plaza J, Rodríguez de Alba M. Fetal genotyping in maternal blood by digital PCR: Towards NIPD of monogenic disorders independently of parental origin. PLoS One. 2016; 11(4): e0153258. doi: 10.1371/journal.pone.0153258. eCollection 2016.
- ↑Zhou L, Zhang B, Liu J, Shi Y, Wang J, Yu B. The optimal cutoff value of Z-scores enhances the judgment accuracy of noninvasive prenatal screening. Front Genet. 2021; 12: 690063. doi: 10.3389/fgene.2021.690063. eCollection 2021.
- ↑Gu W, Koh W, Blumenfeld YJ et al. Noninvasive prenatal diagnosis in a fetus at risk for methylmalonic acidemia. Genet Med. 2014; 16(7): 564-567. doi: 10.1038/gim.2013.194.
- ↑Bustamante-Aragonés A, Rodríguez de Alba M, Perlado S et al. Non-invasive prenatal diagnosis of single-gene disorders from maternal blood. Gene. 2012; 504(1): 144-149. doi: 10.1016/j.gene.2012.04.045.
Paloma Ropero
Servicio de Hematología. Hospital Clínico San Carlos
C/ Profesor Martín Lagos s/n · 28040 Madrid
Tlf.: +34 913 303 321 | E-Mail: paloma.ropero@salud.madrid.org
Año 2024 · número 141 (01) · páginas 19-27
Enviado: 08.12.23
Revisado: 14.12.23
Aceptado: 17.12.23