

Resumen
Introducción: Las hemoglobinopatías representan el grupo más frecuente de enfermedades monogénicas a nivel mundial y su diagnóstico constituye un desafío creciente en contextos de alta diversidad demográfica. Las metodologías convencionales pueden resultar insuficientes para detectar variantes raras, combinaciones multigénicas o alteraciones moduladoras del fenotipo. La secuenciación masiva (Next-generation sequencing, NGS) ha emergido como una herramienta clave para un abordaje diagnóstico integral.
Material y métodos: Se realizó un estudio observacional, descriptivo y retrospectivo que incluyó 1.200 pacientes consecutivos remitidos a un centro de referencia nacional entre octubre de 2024 y noviembre de 2025. Todas las muestras fueron analizadas mediante un panel NGS dirigido a los genes HBA1, HBA2, HBB, HBD, HBG1 y HBG2. Los resultados moleculares se integraron con los datos hematológicos y electroforéticos previos, aplicando las guías ACMG/AMP para la clasificación de variantes.
Resultados: El 80 % de los pacientes presentó alguna alteración molecular en los genes globínicos. Las mutaciones en HBB fueron las más frecuentes (34,4 %), seguidas de las alteraciones en HBA1/HBA2 (31,6 %). Se identificaron combinaciones multigénicas en el 13,3 % de los casos, predominantemente alfa–beta, con un impacto modulador relevante sobre el fenotipo hematológico. La NGS permitió identificar variantes estructurales raras, deleciones extensas, deltapatías y alteraciones en genes gamma, así como resolver múltiples discordancias fenotipo–genotipo y corregir diagnósticos preliminares basados exclusivamente en técnicas convencionales.
Conclusiones: La implementación de NGS multigénica proporciona un incremento sustancial del rendimiento diagnóstico en hemoglobinopatías, especialmente en poblaciones heterogéneas. Este enfoque permite una caracterización molecular integral, optimiza la interpretación clínica, mejora el consejo genético y posiciona a la NGS como una herramienta esencial en el diagnóstico moderno de estas enfermedades.Abstract
Background: Hemoglobinopathies are the most prevalent monogenic disorders worldwide, and their diagnosis has become increasingly complex in multiethnic populations. Conventional diagnostic methods may fail to detect rare variants, multigenic interactions, or genetic modifiers influencing phenotype. Next-generation sequencing (NGS) offers a comprehensive approach to overcome these limitations.
Methods: We conducted a retrospective, observational study including 1,200 consecutive patients referred to a national reference center between October 2024 and November 2025. All samples were analyzed using a targeted NGS panel covering HBA1, HBA2, HBB, HBD, HBG1, and HBG2. Molecular findings were integrated with hematological and electrophoretic data, and variants were classified according to ACMG/AMP guidelines.
Results: Molecular alterations in globin genes were identified in 80% of patients. HBB mutations were the most frequent (34.4%), followed by alterations in HBA1/HBA2 (31.6%). Multigenic combinations were detected in 13.3% of cases, predominantly involving alpha and beta globin genes, with significant phenotypic modulation. NGS enabled the detection of rare structural variants, large deletions, delta-globin and gamma-globin alterations, and resolved numerous genotype–phenotype discrepancies, leading to correction of preliminary diagnoses based on conventional techniques.
Conclusions: Multigene NGS significantly enhances diagnostic accuracy in hemoglobinopathies, particularly in demographically diverse populations. Its ability to identify complex genetic configurations and modulatory variants supports its integration into routine diagnostic workflows, improving clinical interpretation, genetic counseling, and patient management.Palabras clave: Hemoglobinopatías; NGS; Diagnóstico molecular; Talasemia; Análisis multigénico.
Keywords: Hemoglobinopathies; NGS; Molecular diagnosis; Thalassemia; Multigene analysis.
INTRODUCCIÓN
Las hemoglobinopatías constituyen el grupo más prevalente de enfermedades monogénicas a nivel mundial, con una distribución epidemiológica estrechamente ligada a patrones migratorios históricos y contemporáneos [1]. Estas enfermedades incluyen, de forma predominante, los síndromes talasémicos —resultantes de una producción insuficiente de cadenas globínicas alfa o beta— y las variantes estructurales de hemoglobina, como HbS, HbC, HbE o variantes ultra raras con impacto variable en la estabilidad de la proteína [2].
En Europa y, de manera destacada, en países con alta diversidad demográfica, el diagnóstico de hemoglobinopatías ha pasado de ser una tarea relativamente estandarizada a un desafío clínico creciente. La amplia variabilidad fenotípica, las combinaciones genéticas complejas (como la doble heterocigosis α/β) y la existencia de variantes silenciosas o moduladoras hacen que las técnicas tradicionales (High-Performance Liquid Chromatography HPLC, electroforesis capilar, PCR dirigida, Multiplex Ligation-dependent Probe Amplification MLPA) resulten insuficientes para detectar con precisión todas las alteraciones relevantes [3].
La irrupción de la secuenciación masiva (Next-Generation Sequencing NGS) ha supuesto un cambio paradigmático. Esta tecnología permite analizar simultáneamente múltiples genes relacionados con la síntesis y regulación de la hemoglobina —incluyendo regiones codificantes, intrónicas profundas y elementos reguladores—, aumentando drásticamente la capacidad diagnóstica frente a los métodos convencionales [4]. Además, facilita la identificación de variantes raras o combinadas, así como variantes de significado incierto (VUS), que con métodos tradicionales permanecerían sin diagnosticar [5].
En este contexto, la implementación de NGS en el laboratorio permite: a) aumentar la sensibilidad del diagnóstico molecular; b) reducir falsos negativos relacionados con deleciones complejas; c) resolver discordancias entre parámetros hematológicos y electroforéticos; d) mejorar la capacidad predictiva sobre la expresión fenotípica; y e) optimizar el consejo genético y las intervenciones clínicas [6].
El presente estudio describe la experiencia del laboratorio de referencia en este tipo de patologías del Hospital Clínico San Carlos de Madrid, en la que se analizaron 1.200 pacientes mediante un panel NGS dirigido a HBA1, HBA2, HBB, HBD, HBG1 y HBG2.
Los objetivos principales fueron: 1) describir la prevalencia de variantes en genes globínicos; 2) evaluar el rendimiento clínico y diagnóstico de la NGS comparado con la práctica convencional; 3) analizar la correlación entre el genotipo y el fenotipo hematológico; y 4) presentar la casuística anual completa, como reflejo de la actividad de un centro de referencia nacional para hemoglobinopatías.
Este estudio constituye uno de los análisis moleculares más amplios realizados en España mediante NGS, integrando una cohorte altamente diversa que permite abordar la complejidad real de las hemoglobinopatías en nuestro entorno.
MATERIAL Y MÉTODOS
El estudio se diseñó como un análisis observacional, descriptivo y retrospectivo que incluyó a 1.200 pacientes consecutivos remitidos a nuestro laboratorio para el diagnóstico de hemoglobinopatías durante el periodo comprendido entre octubre de 2024 y noviembre de 2025. Este tipo de diseño es ampliamente utilizado en estudios de caracterización molecular de hemoglobinopatías en cohortes clínicas reales [7].
La población estudiada reflejó la diversidad demográfica del área sanitaria y de los centros remitentes, que abarcan hospitales generales, unidades de pediatría, consultas de hematología clínica y programas de cribado familiar y neonatal. Se incluyeron tanto pacientes con sospecha clínica de talasemia o variante estructural como casos en los que las alteraciones hematológicas o electroforéticas generaban dudas diagnósticas. Los principales motivos de derivación incluyeron: microcitosis o anemia no explicada en estudio rutinario, fenotipo hematológico discordante (por ejemplo, HbA2 en límite bajo o elevado sin mutación identificada previamente), hallazgos sugestivos en HPLC o electroforesis capilar, cribado neonatal alterado, estudio familiar de portadores y reevaluación de casos previamente estudiados mediante técnicas convencionales con resultado no concluyente.
Dado que el centro actúa como laboratorio nacional de referencia, una proporción relevante de las muestras correspondía a casos complejos o con discordancia fenotipo–genotipo previa, lo que puede haber influido en la elevada tasa de hallazgos moleculares observada.
Para cada muestra remitida se recogieron datos clínicos básicos —edad, sexo, nacionalidad y motivo de solicitud—, integrándolos posteriormente con los resultados hematimétricos. Los parámetros evaluados (Hb, VCM, HCM y ADE) constituyen la base del algoritmo diagnóstico inicial recomendado para anemias congénitas y hemoglobinopatías [8].
Las fracciones de hemoglobina (HbA2, HbF y variantes estructurales) fueron determinadas mediante HPLC y electroforesis capilar, técnicas reconocidas como estándar de referencia para el cribado inicial de hemoglobinopatías durante la última década [9].
La extracción del ADN genómico se realizó utilizando el sistema automatizado BioRobot EZ1 (Qiagen GmbH, Hilden, Germany), siguiendo las especificaciones del fabricante. La cuantificación se efectuó en un Qubit 4 (Thermo Fisher Scientific, Waltham, MA, USA) con el kit Qubit™ dsDNA High Sensitivity Assay, un método fluorimétrico ampliamente validado para asegurar la pureza y concentración del ADN previo al análisis molecular [10].
El ADN obtenido se sometió a secuenciación masiva mediante el panel Devyser Thalassemia NGS (Devyser, Stockholm, Sweden), diseñado específicamente para el análisis integral de hemoglobinopatías. La aplicabilidad y eficiencia de paneles NGS dirigidos para hemoglobinopatías han sido demostradas en diversos estudios recientes, que destacan su alta sensibilidad para detectar SNV, indels y deleciones/duplicaciones en genes globínicos [3,4,11].
El panel incluía secuencias completas de HBA1, HBA2, HBB, HBD, HBG1 y HBG2, abarcando regiones codificantes y no codificantes con relevancia clínica. La incorporación simultánea de genes alfa, beta, delta y gamma responde a la necesidad creciente de identificar combinaciones multigénicas y variantes moduladoras del fenotipo [12].
El análisis bioinformático se realizó mediante AmpliconSuite v3.2, una plataforma especializada diseñada para el análisis de paneles de amplicones. La clasificación de variantes se llevó a cabo siguiendo las guías ACMG/AMP, estándares internacionales ampliamente adoptados en el diagnóstico molecular [5].
Debido al elevado grado de homología entre HBA1 y HBA2, cualquier variante detectada en regiones susceptibles de asignación ambigua fue confirmada mediante secuenciación Sanger, ya que las regiones con alta similitud de secuencia son propensas a errores técnicos. Numerosos estudios han demostrado que los hallazgos deben confirmarse mediante técnicas independientes basadas en principios distintos [13].
Asimismo, las grandes deleciones del clúster alfa y beta fueron identificadas inicialmente mediante el análisis de profundidad de lectura (read depth) generado por el panel NGS. Posteriormente, estas alteraciones estructurales fueron confirmadas mediante MLPA, técnica considerada estándar de referencia para la validación de deleciones y duplicaciones en los clústeres globínicos [14].
Como parte de nuestras medidas de control de calidad, el laboratorio participa de forma continuada en programas internacionales de control externo específicos para hemoglobinopatías (UK NEQAS) y aplica controles internos y externos en todas las técnicas, garantizando la fiabilidad y trazabilidad de los resultados obtenidos (UK NEQAS Haemoglobinopathy scheme reports, 2018-2024. https://ukneqas.org.uk).
Finalmente, el rendimiento diagnóstico del panel se evaluó integrando los hallazgos moleculares con los datos hematológicos y electroforéticos previos. Se consideró que el NGS aportaba un beneficio clínico cuando permitía resolver discordancias fenotipo–genotipo, aclarar HbA2 límite, identificar persistencias hereditarias de HbF, detectar combinaciones alfa/beta complejas o corregir diagnósticos previos basados únicamente en hematimetría o técnicas bioquímicas. Este enfoque combinó la potencia analítica del NGS con la práctica clínica real, permitiendo una caracterización precisa y comprensiva del perfil genético de las hemoglobinopatías en una cohorte amplia y diversa [15].
RESULTADOS
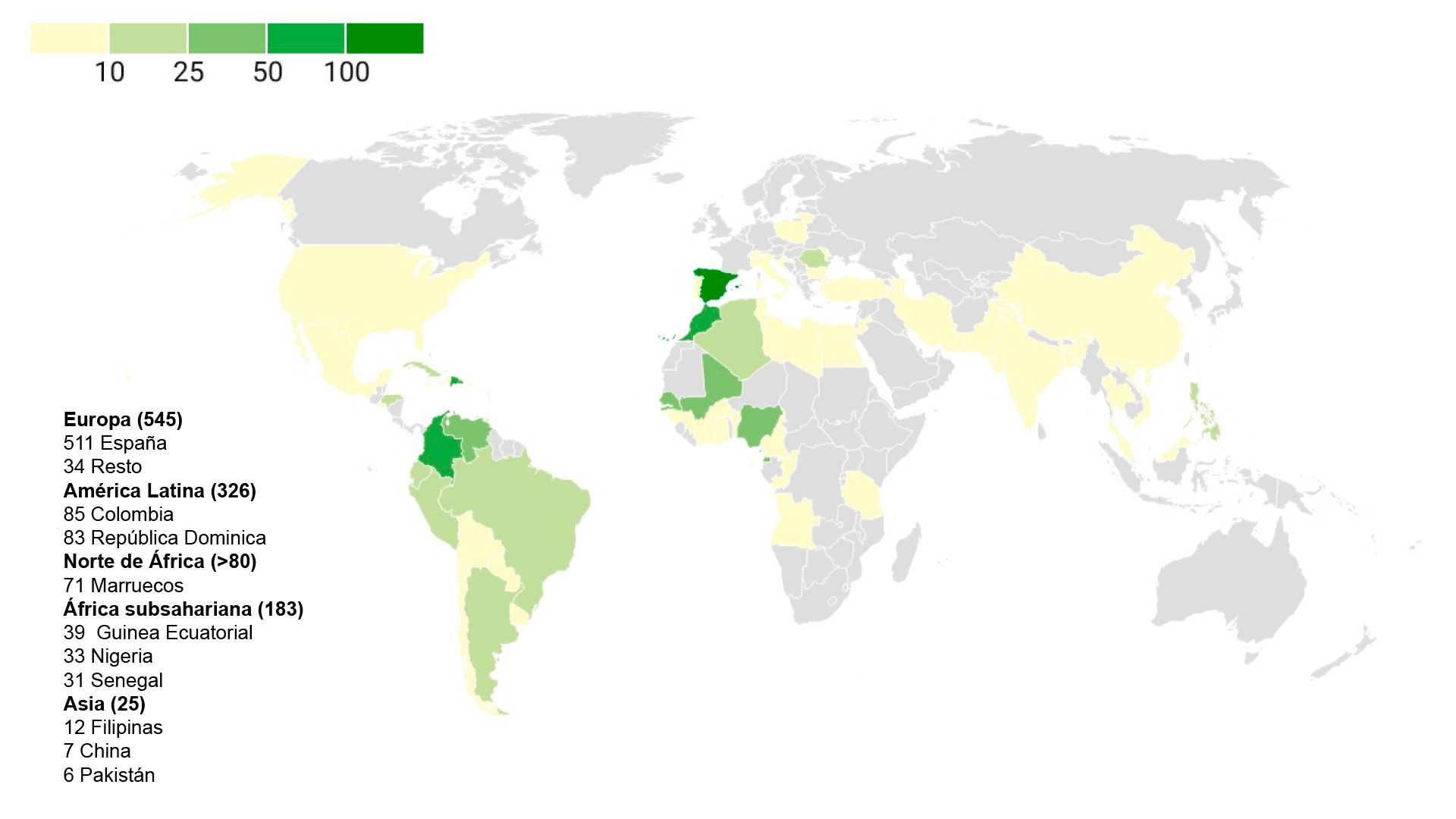
La cohorte estuvo constituida por 1.200 pacientes, con una edad media de 33,4 ± 22,1 años (rango: 0–100). La distribución por sexo fue equilibrada, con un 51,8 % de mujeres y un 46,6 % de varones. En términos de procedencia, la población analizada mostró una marcada heterogeneidad geográfica. En 41 casos (3,4 %) no se disponía de información sobre el país de origen en el momento del análisis. Entre los 1.159 pacientes con procedencia conocida, 511 eran de origen español, 34 europeos y 326 procedían de distintos países de América Latina, principalmente Colombia (n=85) y República Dominicana (n=83). Asimismo, se incluyeron más de 80 pacientes del norte de África, casi todos procedentes de Marruecos (n=71), y 183 de África subsahariana, destacando los casos provenientes de Guinea Ecuatorial (n=39), Nigeria (n=33) y Senegal (n=31), entre otros. Además, se identificaron 19 pacientes de origen asiático, principalmente procedentes de Filipinas (n=12), China (n=7) y Pakistán (n=6) (Figura 1). Esta diversidad se reflejó directamente en el espectro de variantes moleculares identificadas.
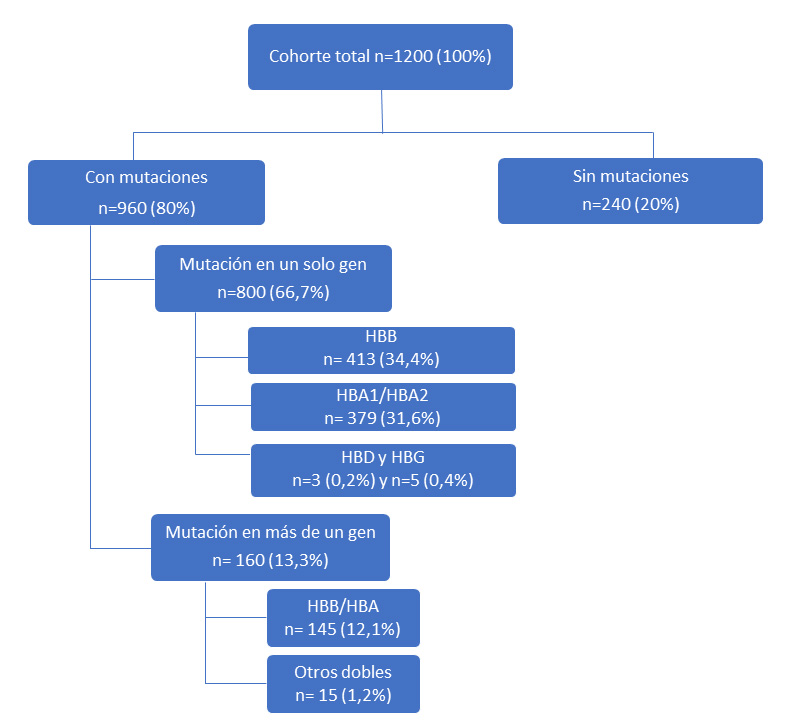
Un total de 240 pacientes (20 %) no presentaron mutaciones en ninguno de los genes analizados (HBA1, HBA2, HBB, HBD, HBG1 y HBG2). En consecuencia, el 80 % de la cohorte (n=960) mostró alguna alteración molecular en los genes globínicos estudiados, siendo las mutaciones en el gen HBB las más frecuentes, identificándose en 413 casos, seguidas de las alteraciones en los genes HBA1/HBA2, presentes en 379 pacientes. Las mutaciones en los genes HBD (n=3) y HBG (n=5) fueron poco frecuentes.
Se identificaron 160 pacientes (13,3 %) con alteraciones simultáneas en dos genes globínicos. La combinación más común correspondió a HBB + HBA (n=145), lo que representa un hallazgo particularmente relevante por su impacto clínico. También se observaron combinaciones menos frecuentes entre HBB + HBD (n=4), HBB + HBG (n=4), HBA + HBG (n=4) y HBA + HBD (n=3) (Figura 2).
Variantes en el gen HBB
Se identificaron mutaciones en el gen HBB en 413 pacientes (34,4 %). De estos, 388 presentaban una única alteración y 25 mostraban dos mutaciones, configurando genotipos compuestos de especial relevancia clínica.
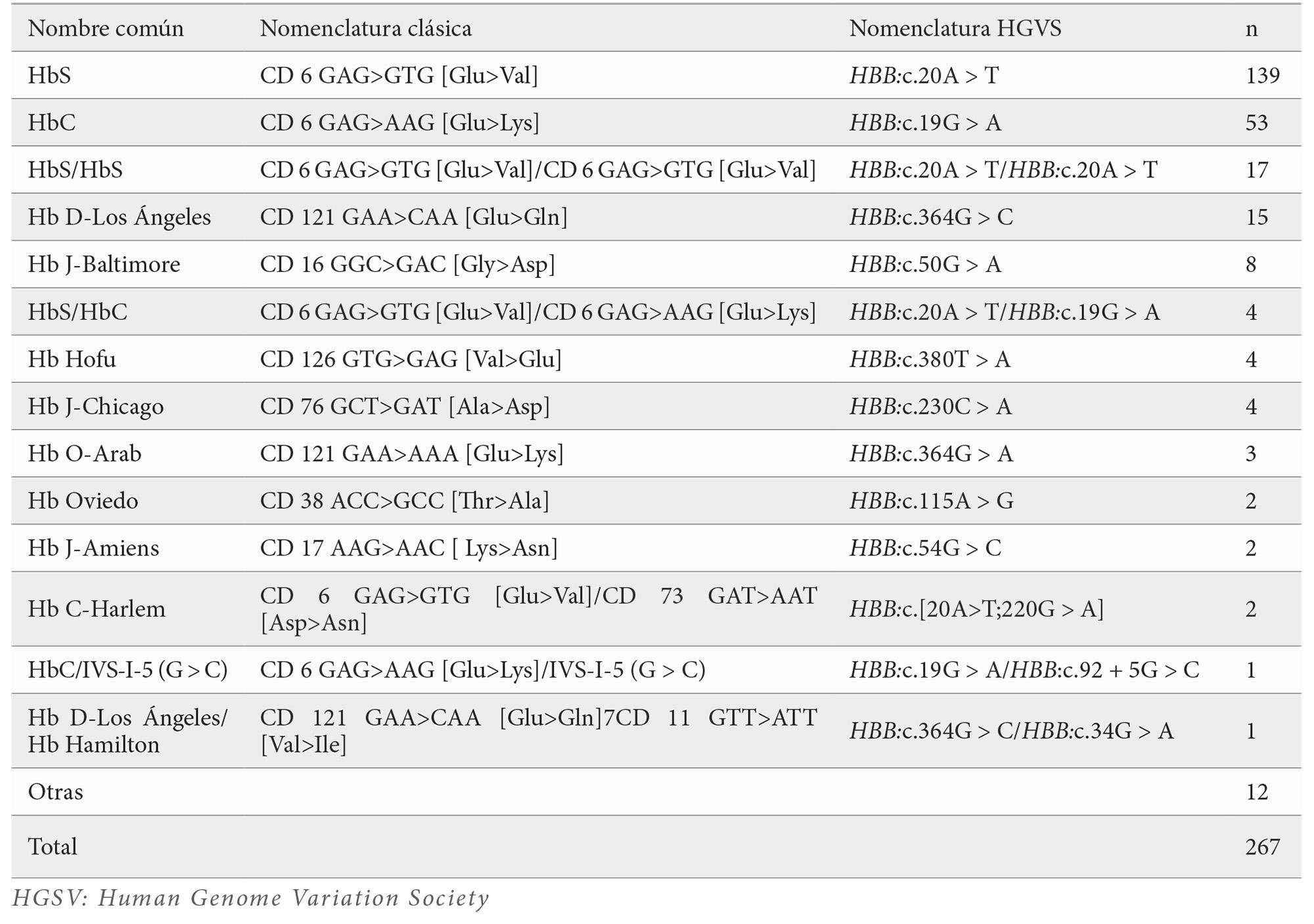
Las hemoglobinopatías estructurales constituyeron el grupo mayoritario entre los portadores de una sola mutación (267 casos). La variante más frecuente fue HbS (n=139), seguida de HbC (n=53) y Hb D–Los Ángeles (n=15). Variantes menos frecuentes, pero representativas de la mezcla étnica de la cohorte, incluyeron Hb J-Baltimore, Hb Hofu, Hb J-Chicago, Hb O-Arab, Hb Oviedo y Hb C-Harlem (Tabla 1). La distribución de HbS y HbC coincidió con los patrones epidemiológicos conocidos en poblaciones de ascendencia africana (HbS: 38%; HbC: 24,7%), afrocaribeña (HbS: 22%; HbC: 28,4%) y sudamericana (HbS: 32%; HbC: 38%).
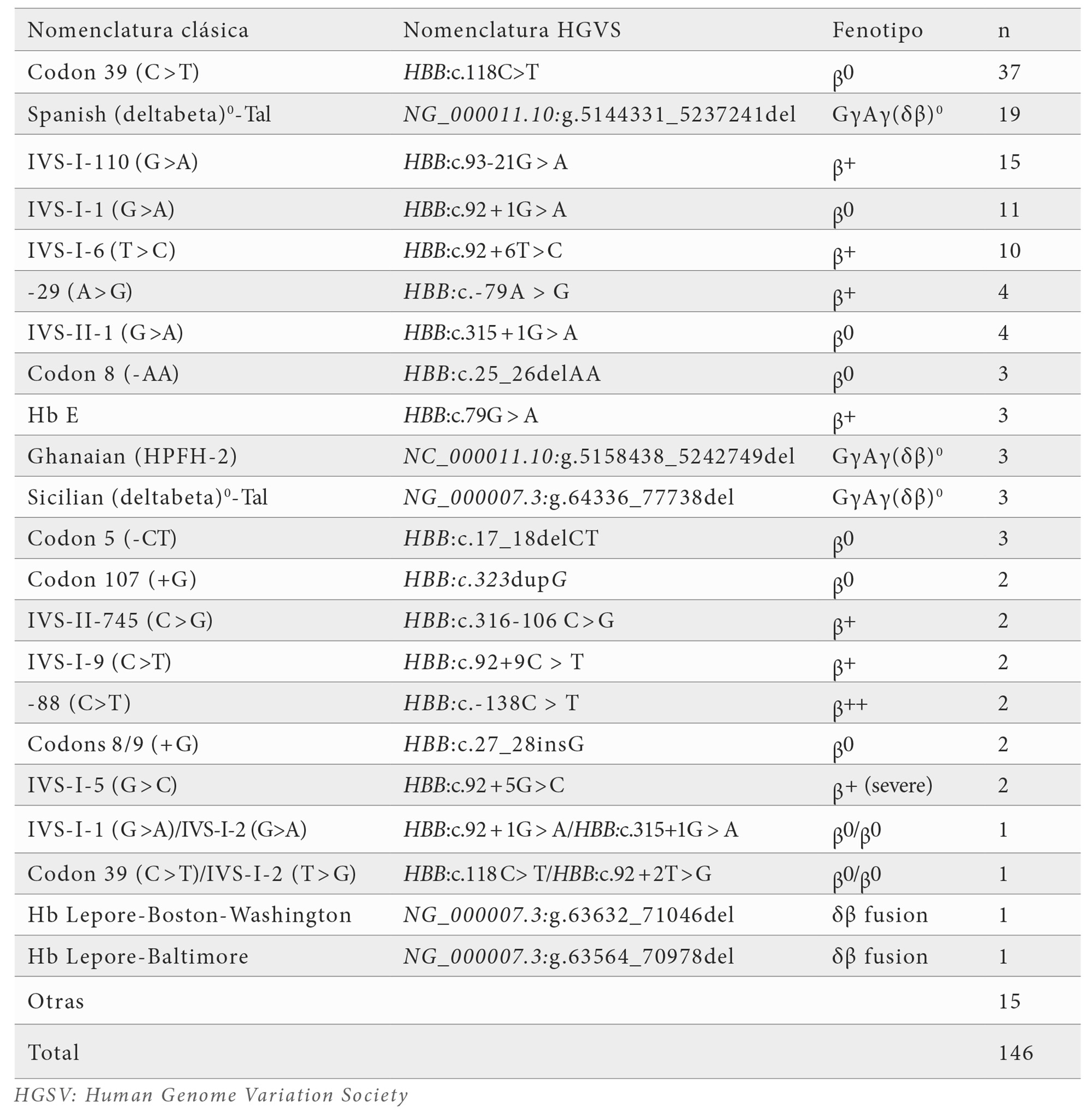
Las beta-talasemias constituyeron un grupo importante (146 casos). Las β⁰-talasemias (n=72) estuvieron dominadas por la mutación Codon 39 (C>T) (n=37), aunque también se identificaron IVS-1-nt1 (G>A), Codon 5 (−CT) y Codon 8 (−AA) y otras variantes graves. Las β⁺ talasemias (n=45) mostraron predominio de las mutaciones IVS-I-nt110 (G>A) e IVS-I-nt6 (T>C). También se registraron 22 casos de δβ⁰ talasemia (deleciones Spanish y Sicilian), 3 casos de HPFH y variantes menos frecuentes como Hb Lepore (Baltimore o Boston) (Tabla 2).
Dentro del grupo de genotipos compuestos de HBB (n=25), se identificaron configuraciones clínicamente relevantes, entre ellas HbS/HbS (anemia falciforme homocigota; n=17); HbS/HbC (n=4); combinaciones doblemente estructurales (Hb D–Los Ángeles/Hb Hamilton) y doblemente heterocigotas de beta talasemia [IVS-1-nt1(G>A)/IVS-2-nt1(G>A) y Codon 39(C>T)/IVS-1-nt2 (G>A)]. Estas últimas, debido a la coexistencia de dos alelos β⁰, generan un fenotipo de β-talasemia mayor.
Variantes en los genes HBA1 y HBA2
Se detectaron 379 pacientes con mutaciones en los genes HBA1 y HBA2, reflejando un elevado grado de alteraciones en el clúster alfa.
La -α3.7/αα, responsable de α⁺ talasemia, fue la variante más prevalente, identificada en 198 pacientes en heterocigosis y 67 en homocigosis (-α3.7/-α3.7α). También se detectaron deleciones -α4.2/αα (n=3) y -α5.2/αα (n=2). Las deleciones α⁰ estuvieron presentes en 30 pacientes, destacando las variantes –SEA (n=14) y –FIL (n=6), típicas del sudeste asiático, así como la deleción –MED1, –KOL y otras macrodeleciones de hasta 212 kb y HS40. Esta última afecta al elemento regulador eritroide principal del clúster α (HS40), cuya pérdida produce una disminución marcada de la expresión de HBA1 y HBA2, funcionalmente equivalente a una deleción α⁰ al suprimir por completo la síntesis de cadenas α en el alelo afectado. Las alfa talasemias no deleción sumaron 35 casos, incluyendo α-5nt (HpH) (n=11), mutaciones en 5’UTR (n=7), alteraciones en regiones 3’UTR, PolyA1 y variantes puntuales distribuidas en diferentes posiciones del gen (Tabla 3).
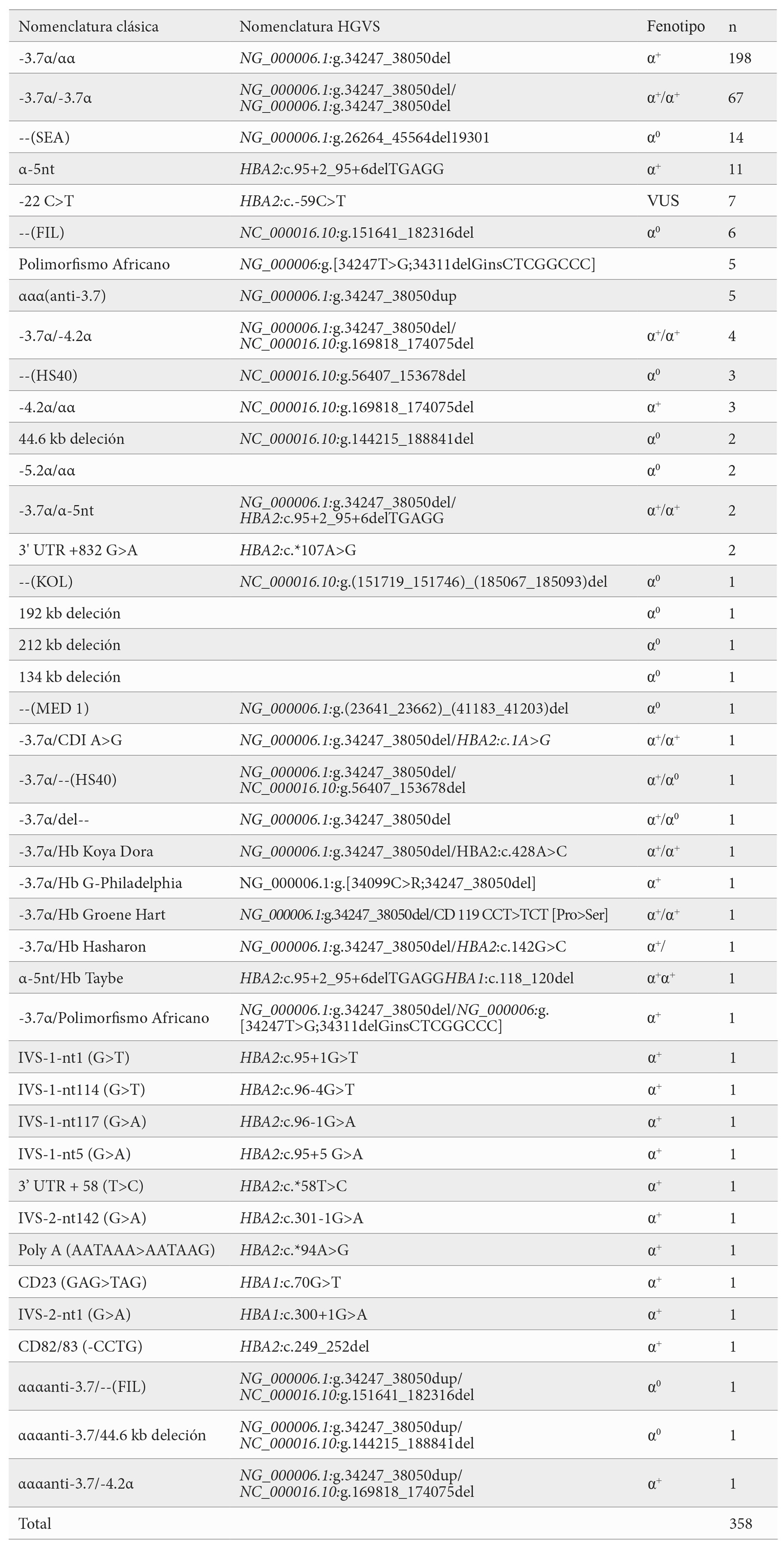
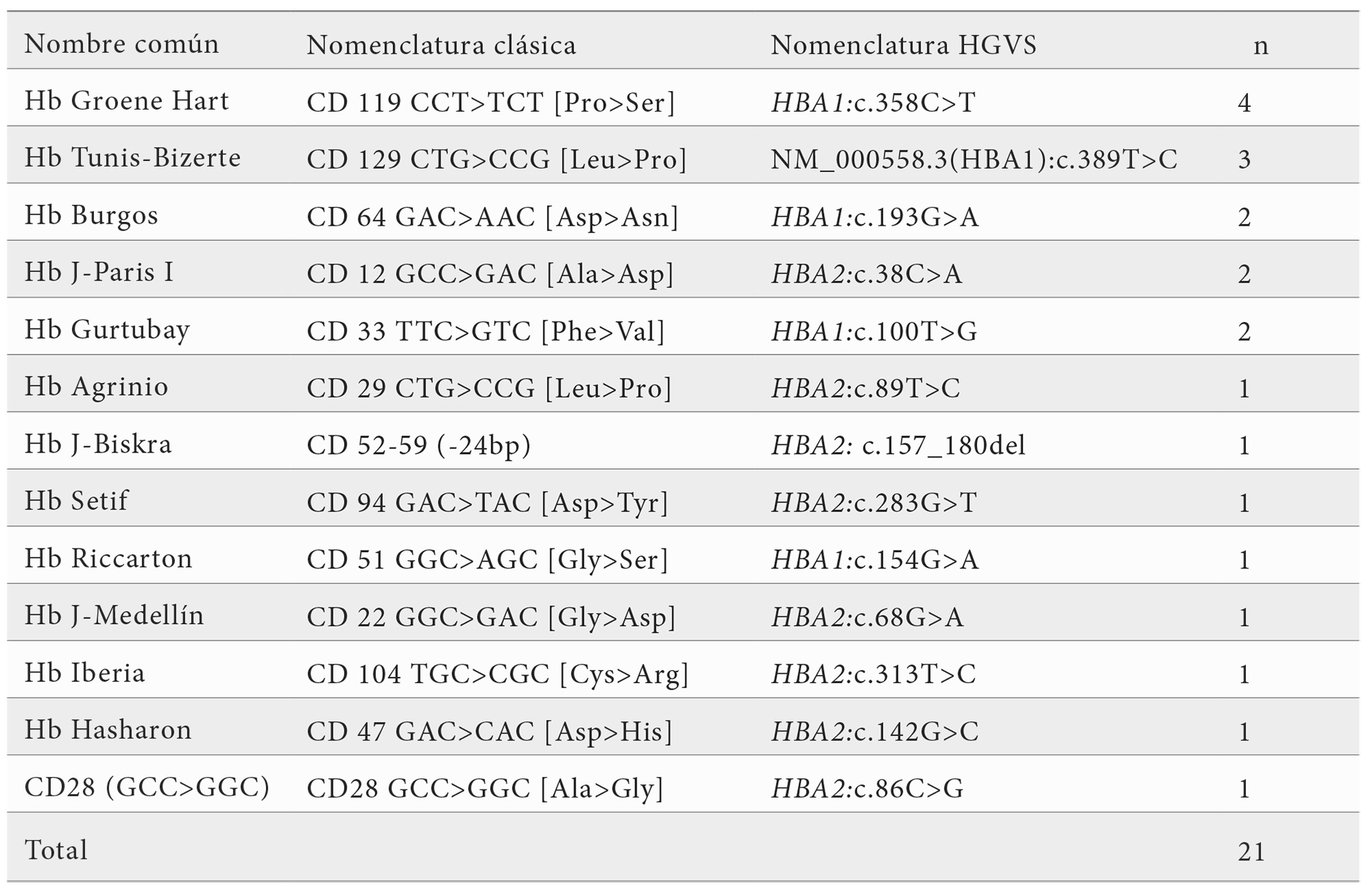
Dentro de las variantes estructurales de cadena alfa se identificaron 21 casos, que incluyeron hemoglobinopatías como Hb Groene Hart, Hb Burgos, Hb Agrinio, Hb Setif, Hb Tunis–Bizerte, Hb Hasharon, Hb J-Medellín o Hb Riccarton. Estas variantes, aunque descritas originalmente en regiones geográficas diversas (Europa, Mediterráneo, norte de África o América Latina), aparecen hoy día en poblaciones heterogéneas como la analizada, reflejo de la movilidad demográfica y de la mezcla étnica de la cohorte (Tabla 4).
Dentro de las variantes detectadas en los genes HBA se identificó el denominado polimorfismo africano, que no representa una mutación patogénica, sino un fenómeno de reconversión génica entre HBA1 y HBA2. Este polimorfismo se asoció predominantemente a pacientes de origen subsahariano y se observó también formando parte de combinaciones multigénicas (por ejemplo, polimorfismo africano + HbS o HbC). Esta reconversión genera un patrón de secuencia híbrido sin impacto funcional, pero con relevancia diagnóstica, ya que puede confundirse con variantes patológicas si no se interpreta en el contexto adecuado.
Desde el punto de vista técnico, se identificó mediante NGS como un patrón de reconversión génica parcial entre HBA1 y HBA2, caracterizado por la presencia de variantes específicas de uno de los genes en el contexto del otro, sin alteración en la profundidad de lectura. Al no implicar variaciones en el número de copias génicas, no es detectable mediante MLPA, que mostró un perfil normal en todos los casos analizados. Los hallazgos fueron confirmados mediante secuenciación Sanger, que evidenció el patrón de reconversión en las regiones diferenciales entre HBA1 y HBA2 (Figura Suplementaria 1).
Este patrón puede simular una alteración de número de copias en determinados amplicones del clúster alfa cuando se interpreta exclusivamente mediante algoritmos automáticos de análisis, por lo que requiere revisión experta e integración con técnicas confirmatorias para evitar interpretaciones erróneas.
Asimismo, se identificaron 8 pacientes con triplicaciones del gen alfa. De ellas, cuatro estaban asociadas a mutaciones en el gen HBB, formando combinaciones alfa–beta de especial interés clínico. En estos, la triplicación actuó como un modulador fenotípico, intensificando la microcitosis y agravando el fenotipo en portadores heterocigotos de β-talasemia, lo que subraya su relevancia diagnóstica. Las restantes triplicaciones correspondieron a reordenamientos alfa aislados sin alteraciones beta concomitantes.
Variantes en HBD y HBG
Aunque menos frecuentes, las variantes detectadas en HBD (n=3) y HBG (n=5) resultaron esenciales para la correcta interpretación de perfiles hematológicos. Las mutaciones en HBD justificaron valores de HbA2 inesperadamente bajos o en límite bajo de la normalidad, evitando confundirlos con patrones atípicos de β-talasemia. Por su parte, las variantes en la región reguladora −158 de HBG justificaron los incrementos de HbF observados en pacientes que no presentaban mutaciones capaces de reducir la producción de β-globina.
Combinaciones multigénicas
Un hallazgo destacado del estudio fue la identificación de 160 pacientes (13,3 %) con alteraciones simultáneas en dos genes globínicos, proporción superior a la descrita en series convencionales y que justifica el uso de un enfoque NGS multigénico (Tabla 5).
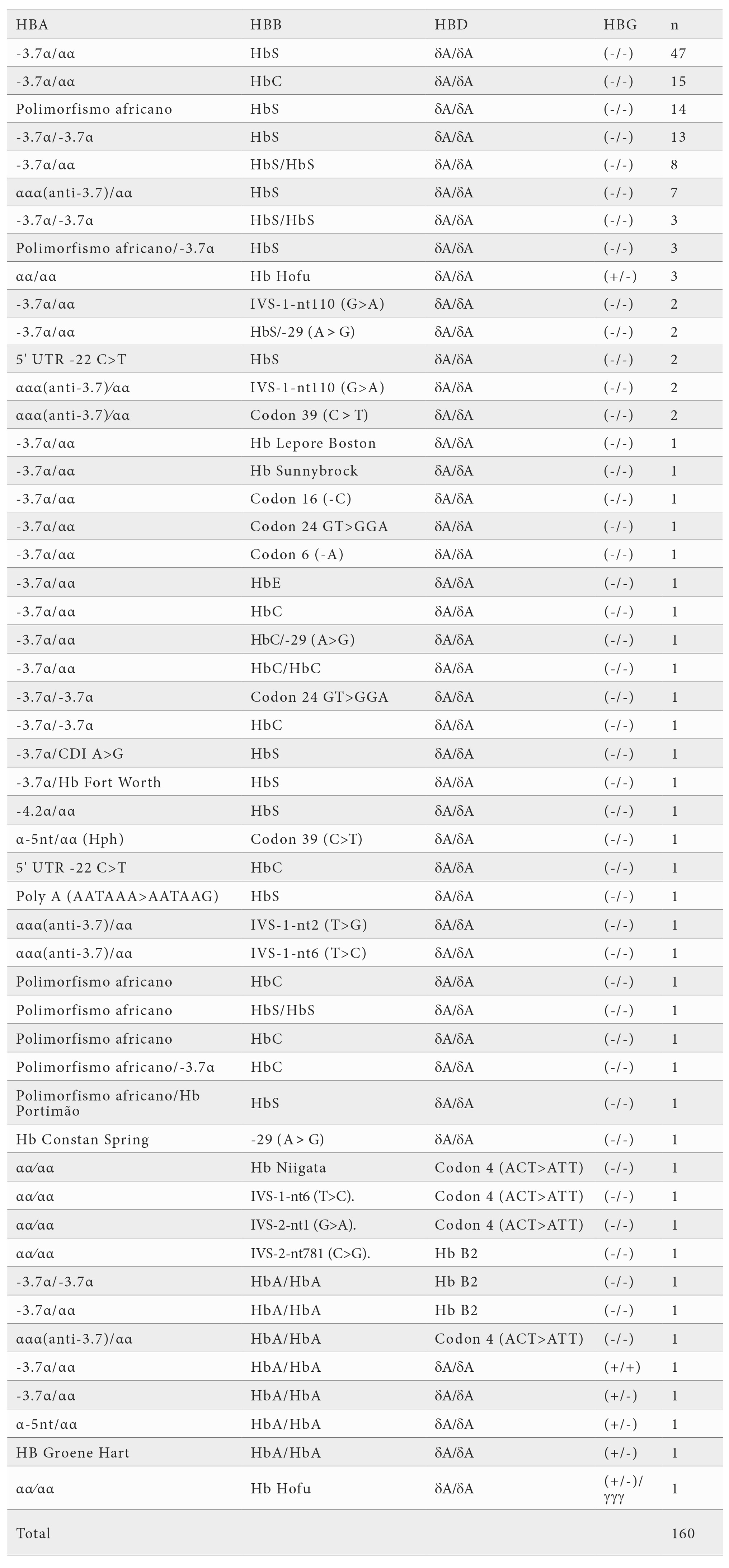
Las combinaciones HBB + HBA constituyeron el grupo predominante. Se identificaron 47 pacientes con HbS + -α3.7/αα; 15 con HbC + -α3.7/αα, así como casos que combinaban beta-talasemias con variantes alfa no deleción o con triplicaciones alfa. Estas configuraciones evidenciaron un impacto modulador significativo sobre el fenotipo, ya que alteran parámetros como el VCM, la concentración intracelular de hemoglobinas anómalas o la proporción de HbA2.
Además, se observaron combinaciones menos frecuentes, pero clínicamente relevantes entre HBB y HBD (n=4), HBB y HBG (n=3) y HBA y HBG (n=4). Estas interacciones explicaron perfiles hematológicos no concluyentes, como HbA2 en el límite bajo de la normalidad o HbF inesperadamente elevada.
El análisis mediante NGS permitió resolver múltiples discordancias fenotipo–genotipo detectadas inicialmente por hematimetría o electroforesis, especialmente en pacientes con microcitosis no explicada, valores de HbA2 no concluyentes o niveles de HbF inesperadamente elevados. En los casos con HbA2 en el límite bajo de la normalidad, el panel permitió identificar deltapatías, δβ⁰ talasemias o combinaciones alfa–beta que justificaban patrones electroforéticos ambiguos.
En varios pacientes, estos hallazgos moleculares condujeron a corregir diagnósticos preliminares establecidos exclusivamente con parámetros hematológicos o electroforéticos, permitiendo establecer el genotipo definitivo.
El análisis simultáneo de los cuatro loci globínicos permitió reconstruir de forma integral las configuraciones genotípicas, identificar efectos aditivos y moduladores y evitar errores diagnósticos propios de aproximaciones parciales. No se detectaron combinaciones que afectaran de manera concomitante a tres o más genes.
DISCUSIÓN
El presente estudio proporciona una visión exhaustiva del espectro molecular de las hemoglobinopatías en una cohorte amplia y marcadamente heterogénea, evaluada íntegramente mediante un panel NGS dirigido a HBA1, HBA2, HBB, HBD, HBG1 y HBG2. Esta aproximación permitió no solo caracterizar un volumen considerable de variantes estructurales y talasémicas, sino también identificar un número significativo de configuraciones multigénicas, cuya relevancia clínica se ha infravalorado tradicionalmente debido a las limitaciones de las metodologías convencionales [3,4,6]. Además, este estudio constituye uno de los análisis moleculares más amplios realizados en el ámbito español en la era post–NGS, incorporando una cohorte altamente diversa que refleja con precisión la movilidad demográfica contemporánea y su impacto en el espectro genético de las hemoglobinopatías.
La diversidad geográfica y étnica de la cohorte analizada —con predominio de pacientes procedentes de España, el Caribe, Sudamérica y África occidental— confiere a este estudio una representatividad singular dentro del contexto europeo, donde los patrones migratorios han transformado de forma sustancial el perfil genético de las hemoglobinopatías [16].
Desde un punto de vista epidemiológico, las variantes identificadas reproducen fielmente las distribuciones geográficas conocidas, la elevada prevalencia de HbS y HbC en pacientes procedentes de regiones con ascendencia africana o afrocaribeña; la presencia de β-talasemia de origen mediterráneo (como Codon 39 o delecciones δβ⁰ Spanish y Sicilian) en pacientes españoles y norteafricanos; y la detección de deleciones alfa típicas en población española y áreas africanas (-α3.7/αα) o asiáticas (–SEA, –FIL) [16,17,18]. Estos hallazgos refuerzan la necesidad, ya reflejada en estudios europeos recientes, de que la composición poblacional actual exige metodologías diagnósticas que aborden simultáneamente variantes con orígenes y comportamientos clínicos muy distintos, lo que justifica plenamente la adopción de paneles NGS amplios [4,19].
Uno de los elementos diferenciales de este trabajo es la identificación sistemática de un número considerable de combinaciones multigénicas, presentes en 160 pacientes (13,3 %). Este porcentaje, muy superior al que comunican series diagnósticas basadas únicamente en métodos bioquímicos o aproximaciones secuenciales, subraya la complejidad genética real de las hemoglobinopatías en nuestro entorno [3,4].
La elevada proporción de configuraciones multigénicas observada en nuestra cohorte debe interpretarse asimismo en el contexto del perfil de derivación del centro. Como laboratorio nacional de referencia, recibimos un número significativo de casos complejos, incluidos pacientes con fenotipos discordantes, estudios previos no concluyentes o sospecha de interacción genética múltiple. Este sesgo de selección puede haber contribuido tanto a la alta tasa global de positividad molecular (80 %) como a la frecuencia de combinaciones alfa–beta superior a la descrita en series poblacionales no seleccionadas. En este sentido, nuestros resultados reflejan la casuística propia de un centro de alta especialización más que la de una cohorte poblacional general.
Aunque no fue posible cuantificar retrospectivamente el número exacto de casos remitidos específicamente por discrepancia genotipo–fenotipo documentada, el análisis de los motivos de derivación y la experiencia clínica del laboratorio indican que una proporción relevante correspondía a situaciones diagnósticas no concluyentes, microcitosis no explicadas o perfiles electroforéticos ambiguos. En estos contextos, el análisis multigénico mediante NGS permitió establecer el genotipo definitivo y esclarecer la correlación fenotípica en un número significativo de pacientes.
Las combinaciones alfa–beta representaron el grupo predominante, como ya se ha descrito en estudios que documentan la interacción moduladora de las deleciones alfa sobre el fenotipo de portadores de HbS o β-talasemia [20,21,22]. La identificación de interacciones entre HBB y HBD, HBB y HBG, o HBA y HBG coincide igualmente con la literatura, que destaca cómo estas variantes pueden dar lugar a perfiles electroforéticos ambiguos (HbA2 en el límite bajo de la normalidad, HbF elevada) que requieren un enfoque multigénico para su correcta interpretación [23,24].
En este contexto, un aspecto técnico particularmente relevante es la dificultad inherente al análisis molecular de HBA1 y HBA2, debido al elevado grado de homología entre ambos genes [25]. Aunque el panel NGS utilizado permitió detectar de forma robusta variantes puntuales, indels y deleciones, la asignación inequívoca de mutaciones localizadas en regiones compartidas requirió confirmación mediante secuenciación Sanger. Esta necesidad ha sido ampliamente documentada en estudios que analizan la arquitectura duplicada del clúster α y sus implicaciones diagnósticas [13]. Esta limitación técnica refuerza la importancia de integrar métodos complementarios cuando se estudian regiones altamente homólogas, incluso en un contexto NGS de alto rendimiento [13,26].
Un aspecto relevante del análisis de los genes alfa es la presencia del denominado polimorfismo africano, que no corresponde a una mutación patogénica, sino a una reconversión génica parcial entre HBA1 y HBA2. Este proceso de gene conversion es característico de poblaciones de origen africano y carece de efecto clínico, pero su identificación es esencial para evitar interpretaciones erróneas, especialmente en individuos con microcitosis o en combinaciones multigénicas donde podría confundirse con una variante alfa no deleción [27].
Diversos estudios han demostrado que la coexistencia de deleciones alfa con variantes estructurales como HbS o HbC puede modificar de manera sustancial parámetros hematimétricos como el VCM o la concentración intracelular de las hemoglobinas anómalas, atenuando o exacerbando determinados rasgos clínicos [20,21,28]. De forma análoga, se ha descrito que la presencia de triplicaciones alfa en portadores de β-talasemia heterocigota se asocia con una mayor gravedad del fenotipo [22], mientras que variantes en los genes de la γ-globina (HBG) explican niveles elevados de HbF en pacientes que, a priori, no presentan mutaciones depresoras en el gen HBB [24].
En nuestra cohorte, la elevada frecuencia de combinaciones de mutaciones en HBA y HBB, así como la identificación de alteraciones en los genes de la γ-globina, es coherente con este modelo de interacción genética. Estos hallazgos subrayan la necesidad de interpretar los resultados dentro del contexto del genoma globínico completo y no desde un único gen, un principio ampliamente destacado en estudios recientes que abogan por el análisis integral del clúster globínico [4,19].
La utilidad clínica del enfoque multigénico empleado es especialmente evidente en situaciones en las que el fenotipo observado se desvía de lo esperado, un fenómeno ampliamente documentado en estudios de correlación genotipo–fenotipo en hemoglobinopatías [5,15]. En este sentido, el panel NGS permitió resolver discordancias fenotipo–genotipo, aclarar perfiles de HbA2 y HbF no concluyentes y corregir diagnósticos preliminares basados únicamente en parámetros hematológicos o bioquímicos. Este beneficio ha sido señalado por varios grupos que comparan el rendimiento de paneles NGS con métodos convencionales [3,4,6,19].
El rendimiento diagnóstico del panel Devyser empleado quedó firmemente demostrado en nuestro estudio. Su capacidad para detectar variantes puntuales, deleciones extensas y alteraciones en genes altamente homólogos posiciona este enfoque como una herramienta de alto valor añadido en entornos clínicos con diversidad demográfica. La incorporación simultánea de HBD y HBG aportó un valor adicional en casos con discrepancias fenotipo–genotipo, permitiendo resolver perfiles electroforéticos atípicos y clarificar la contribución de las deltapatías y variantes gamma en la modulación de HbA2 y HbF, respectivamente [23,24]. Este enfoque contrasta con las limitaciones de HPLC, electroforesis capilar, PCR dirigida o MLPA, técnicas que, si bien continúan siendo esenciales, resultan insuficientes para capturar variantes reguladoras, combinaciones complejas o interacciones moduladoras entre genes.
Los resultados obtenidos se alinean con la creciente evidencia internacional que respalda la implementación de NGS en el diagnóstico rutinario de hemoglobinopatías, especialmente en regiones donde la diversidad de variantes y la movilidad poblacional generan fenotipos atípicos o discordantes [2,4,19]. Estudios recientes publicados en Europa, Norteamérica y Asia muestran una tendencia similar, aunque con diferencias en la distribución mutacional atribuibles a variaciones regionales [3,11,12,29,30]. En este contexto, el presente trabajo constituye, hasta donde alcanza la literatura disponible, uno de los análisis moleculares más completos realizados en España, tanto por el tamaño muestral como por la profundidad diagnóstica alcanzada.
No obstante, el estudio presenta ciertas limitaciones. Aunque el panel NGS permite un análisis amplio, no cubre algunas regiones distales o elementos reguladores cuya contribución fenotípica podría ser significativa en casos seleccionados. En particular, no cubre de forma completa la región reguladora LCR del clúster beta, donde se localizan los sitios hipersensibles HS1–HS5 implicados en la regulación de HbF y algunas formas no delecionales de HPFH. Tampoco incluye los enhancers eritroides de BCL11A, los promotores extendidos y regiones intergénicas profundas del clúster beta (como las zonas upstream de HBG1–HBG2), ni las regiones reguladoras distales del clúster alfa, especialmente el enhancer HS-40, cuya alteración puede modular de forma significativa la expresión de HBA [31,32,33]. Asimismo, mutaciones intrónicas profundas o reordenamientos estructurales complejos pueden escapar a la detección por NGS de panel, lo que refuerza la necesidad de estudios complementarios en casos atípicos [31].
La identificación de variantes de significado incierto (VUS) requiere una interpretación cautelosa, especialmente en ausencia de correlación familiar o funcional. La literatura señala la importancia de integrar predictores in silico, bases de datos internacionales y, cuando es posible, estudios funcionales o familiares para ajustar su clasificación [5,34]. Además, el carácter retrospectivo del estudio y la variabilidad en los motivos de derivación pueden haber generado cierto sesgo en la prevalencia observada de algunas alteraciones, aunque la amplitud de la cohorte contribuye a compensar esta limitación.
A pesar de estas consideraciones, los hallazgos del estudio tienen implicaciones clínicas directas. La caracterización completa del genotipo permite optimizar el consejo genético, orientar estrategias de cribado familiar y prenatal, interpretar de forma más precisa los perfiles hematológicos en el límite de la normalidad y ajustar el seguimiento clínico de portadores con riesgo potencial de complicaciones, aspectos ampliamente reconocidos en la literatura [5,15,20]. Asimismo, la identificación temprana de combinaciones multigénicas facilita la estratificación pronóstica y contribuye a la elección de intervenciones terapéuticas más adecuadas, como el control de factores desencadenantes en portadores de Hb S con variantes moduladoras o la prevención de anemia crónica en casos con interacciones alfa–beta complejas, y podría tener implicaciones relevantes en la interpretación de la respuesta y los resultados de futuras terapias génicas
En conjunto, los resultados de este trabajo refuerzan la necesidad de integrar tecnologías NGS en el abordaje diagnóstico de las hemoglobinopatías, especialmente en contextos de alta heterogeneidad demográfica. La capacidad del panel empleado para detectar variantes múltiples, estructurales y regulatorias, así como configuraciones multigénicas con impacto clínico, sitúa este enfoque como una herramienta esencial en la práctica clínica contemporánea. Futuros estudios multicéntricos podrían ampliar estas observaciones y contribuir a establecer algoritmos diagnósticos estandarizados que combinen de manera óptima las pruebas bioquímicas tradicionales con el análisis genómico de nueva generación [2].
AGRADECIMIENTOS
Rafael Sánchez-del Hoyo de la Unidad de Apoyo Metodológico a la Investigación, Hospital Clínico San Carlos, IdISSC, Madrid.
DECLARACIÓN DE TRANSPARENCIA
Los autores/as de este artículo declaran no tener ningún tipo de conflicto de intereses respecto a lo expuesto en el presente trabajo.
BIBLIOGRAFÍA
- ↑Piel FB, Steinberg MH, Rees DC. Sickle Cell Disease. N Engl J Med. 2017;376(16):1561-1573. doi: 10.1056/NEJMra1510865.
- ↑Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4:18010. doi: 10.1038/nrdp.2018.10.
- ↑Achour A, Koopmann TT, Baas F, Harteveld CL. The Evolving Role of Next-Generation Sequencing in Screening and Diagnosis of Hemoglobinopathies. Front Physiol. 2021;12:686689. doi: 10.3389/fphys.2021.686689.
- ↑Rizzuto V, Koopmann TT, Blanco-Álvarez A, Tazón-Vega B, Idrizovic A, Díaz de Heredia C et al. Usefulness of NGS for Diagnosis of Dominant Beta-Thalassemia and Unstable Hemoglobinopathies in Five Clinical Cases. Front Physiol. 20215;12:628236. doi: 10.3389/fphys.2021.628236.
- ↑Harrison SM, Biesecker LG, Rehm HL. Overview of Specifications to the ACMG/AMP Variant Interpretation Guidelines. Curr Protoc Hum Genet. 2019;103(1):e93. doi: 10.1002/cphg.93.
- ↑Selvatici R, Guida V, Maffei M, Irrera MA, Margutti A, Bisceglia P et al. Relevance of Next-Generation Sequencing in the Diagnosis of Thalassemia and Hemoglobinopathies: The Experience of Four Italian Diagnostic Hubs. Genes 2025, 16, 28. https://doi.org/10.3390/genes16010028.
- ↑Kattamis A, Forni GL, Aydinok Y, Viprakasit V. Changing patterns in the epidemiology of β-thalassemia. Eur J Haematol. 2020;105(6):692-703. doi: 10.1111/ejh.13512. Epub 2020 Sep 21.
- ↑Bain BJ. Haemoglobinopathy diagnosis: algorithms, lessons and pitfalls. Blood Rev. 2011;25(5):205-13. doi: 10.1016/j.blre.2011.04.001. Epub 2011 May 19.
- ↑Gulbis B, Fontaine B, Vertongen F, Cotton F. The place of capillary electrophoresis techniques in screening for haemoglobinopathies. Ann Clin Biochem. 2003;40(Pt 6):659-62. doi: 10.1258/000456303770367261.
- ↑Naidoo ALK, Martin DLP, Heathfield LJ. Establishment of performance parameters for the Qubit™ 1X dsDNA HS Assay for use in forensic genetics casework. Forensic Science International 2025; 11: 100405. DOI pendiente de indexación
- ↑Shang X, Peng Z, Ye Y, Asan, Zhang X, Chen Y, et al. Rapid Targeted Next-Generation Sequencing Platform for Molecular Screening and Clinical Genotyping in Subjects with Hemoglobinopathies. EBioMedicine. 2017;23:150-159. doi: 10.1016/j.ebiom.2017.08.015. Epub 2017 Aug 17.
- ↑Zulkeflee RH, Bahar R, Abdullah M, Mohd Radzi MAR, Md Fauzi A, Hassan R. Application of Targeted Next-Generation Sequencing for the Investigation of Thalassemia in a Developing Country: A Single Center Experience. Diagnostics (Basel). 2023;13(8):1379. doi: 10.3390/diagnostics13081379.
- ↑Aliyeva G, Asadov C, Mammadova T, Gafarova S, Abdulalimov E. Thalassemia in the laboratory: pearls, pitfalls, and promises. Clin Chem Lab Med. 2018;57(2):165-174. doi: 10.1515/cclm-2018-0647.
- ↑Harteveld CL. State of the art and new developments in molecular diagnostics for hemoglobinopathies in multiethnic societies. Int J Lab Hematol. 2014;36(1):1-12. doi: 10.1111/ijlh.12108. Epub 2013 May 31.
- ↑Thein SL. Genotype–phenotype relationships in hemoglobinopathies. Hematology Am Soc Hematol Educ Program. 2013;2013(1):354-361
- ↑Weatherall DJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115(22):4331-6. doi: 10.1182/blood-2010-01-251348. Epub 2010 Mar 16.
- ↑Origa R. β‑Thalassemia. Genet Med. 2017;19(6):609‑619. Epub 2016 Nov 3. DOI: 10.1038/gim.2016.173.
- ↑Higgs DR. The molecular basis of α-thalassemia. Cold Spring Harb Perspect Med. 2013;3(1):a011718. doi: 10.1101/cshperspect.a011718.
- ↑Harteveld CL, Achour A, Arkesteijn SJG, Ter Huurne J, Verschuren M, Bhagwandien-Bisoen S et al. The hemoglobinopathies, molecular disease mechanisms, and diagnostics. Int J Lab Hematol. 2022;44 Suppl 1(Suppl 1):28-36. doi: 10.1111/ijlh.13885.
- ↑Fasola FA, Babalola OA, Brown BJ, Odetunde A, Falusi AG, Olopade O. The Effect of Alpha Thalassemia, HbF and HbC on Haematological Parameters of Sickle Cell Disease Patients in Ibadan, Nigeria. Mediterr J Hematol Infect Dis. 2022;14(1):e2022001. doi: 10.4084/MJHID.2022.001.
- ↑Hamid M, Keikhaei B, Galehdari H, Saberi A, Sedaghat A, Shariati G et al. Alpha-globin gene triplication and its effect in beta-thalassemia carriers, sickle cell trait, and healthy individuals. EJHaem. 2021;2(3):366-374. doi: 10.1002/jha2.262.
- ↑Ropero P, González Fernández FA, Nieto JM, Torres-Jiménez WM, Benavente C. β-Thalassemia Intermedia: Interaction of α-Globin Gene Triplication With β-thalassemia Heterozygous in Spain. Front Med (Lausanne). 2022;9:866396. doi: 10.3389/fmed.2022.866396.
- ↑Harteveld C.L. Diagnosis of Haemoglobinopathies: New Scientific Advances. Thalass. Rep. 2018; 8: 7473. https://doi.org/10.4081/thal.2018.7473
- ↑Sankaran VG, Xu J, Byron R, Greisman HA, Fisher C, Weatherall DJ et al. A functional element necessary for fetal hemoglobin silencing. N Engl J Med. 2011;365(9):807-14. doi: 10.1056/NEJMoa1103070.
- ↑de la Fuente-Gonzalo F, Nieto JM, Villegas A, González FA, Martínez R, Ropero P. Characterization of deletional and non-deletional alpha globin variants in a large cohort from Spain between 2009 and 2014. Ann Hematol. 2019 Apr 25. doi: 10.1007/s00277-019-03696-w
- ↑Mettananda S, Higgs DR. Molecular Basis and Genetic Modifiers of Thalassemia. Hematology/Oncology Clin. North America 2018;32(2):177–191. doi:10.1016/j.hoc.2017.11.003
- ↑Viprakasit V, Ekwattanakit S. Clinical Classification, Screening and Diagnosis for Thalassemia. Hematol Oncol Clin North Am. 2018;32(2):193-211. doi: 10.1016/j.hoc.2017.11.006.
- ↑González FA, Blázquez C, Ropero P, Aláez C, Polo M et al. Asociación de hemoglobinopatía S y alfa-talasemia. Análisis de 45 casos [Association of hemoglobinopathy and alpha thalassemia. Study of 45 patients]. Med Clin (Barc). 2005;124(19):726-9. doi: 10.1157/13075443
- ↑He J, Song W, Yang J, Lu S, Yuan Y, Guo J et al. Next-generation sequencing improves thalassemia carrier screening among premarital adults in a high-prevalence population: the Dai nationality, China. Genet Med. 2017;19(9):1022-1031. doi: 10.1038/gim.2016.218. Epub 2017 Jan 26.
- ↑Quarmyne MO, Bock F, Lakshmanan S, Attell BK, Snyder A, Boudreaux J et al. Newborn Screening for Sickle Cell Disease and Thalassemia. JAMA Health Forum. 2025;6(3):e250064. doi: 10.1001/jamahealthforum.2025.0064.
- ↑Hardison RC. Evolution of hemoglobin and its genes. Cold Spring Harb Perspect Med. 2012;2(12):a011627. doi: 10.1101/cshperspect.a011627. PMID: 23209182; PMCID: PMC3543078
- ↑Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Van Handel B et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science (New York, N.Y.). 2008 Dec;322(5909):1839-1842. DOI: 10.1126/science.1165409.
- ↑Menzel, S., Thein, S.L. Genetic Modifiers of Fetal Haemoglobin in Sickle Cell Disease. Mol Diagn Ther 2019;23: 235–244. https://doi.org/10.1007/s40291-018-0370-8
- ↑Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405-24. doi: 10.1038/gim.2015.30. Epub 2015 Mar 5.
Paloma Ropero
Servicio de Hematología y Hemoterapia. Hospital Clínico San Carlos
C/Profesor Martín Lagos s/n · 28040 Madrid
Tlf.: +34 913 303 000 | E-Mail: paloma.ropero@salud.madrid.org
Enviado: 08.01.26
Revisado: 16.01.26
Aceptado: 28.01.26